
Part of Special Issue:
Synthetic Biology in Therapeutics and Healthcare: Innovations and Applications
Download PDF
Cite This Article
Application of Synthetic Biology to the Biosynthesis of Polyketides
Author Information
Other Information
1
Key Laboratory for Green Processing of Chemical Engineering of Xinjiang Bingtuan, School of Chemistry and Chemical Engineering, Shihezi University, Shihezi 832003, China
2
Academy of National Food and Strategic Reserves Administration, Institute of Cereal & Oil Science and Technology, Beijing 100037, China
3
Key Laboratory of Medical Molecule Science and Pharmaceutics Engineering, Ministry of Industry and Information Technology, Institute of Biochemical Engineering, School of Chemistry and Chemical Engineering, Beijing Institute of Technology, Beijing 100081, China
4
Department of Chemical Engineering, Tsinghua University, Beijing 100084, China
5
Key Lab for Industrial Biocatalysis, Ministry of Education, Tsinghua University, Beijing 100084, China
6
Center for Synthetic and Systems Biology, Tsinghua University, Beijing 100084, China
*
Authors to whom correspondence should be addressed.
Received: 15 July 2024 Accepted: 12 August 2024 Published: 14 August 2024

© 2024 The authors. This is an open access article under the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/).
Synth. Biol. Eng.
2024,
2(3), 10012;
DOI: 10.35534/sbe.2024.10012
ABSTRACT:
Polyketides (PKs) are a large class of secondary
metabolites produced by microorganisms and plants, characterized by highly
diverse structures and broad biological activities. They have wide market and
application prospects in medicine, agriculture, and the food industry. The
complex chemical structures and multiple steps of natural polyketides result in
yield that cannot be met by purely synthetic methods. With the development of
synthetic biology, a number of novel technologies and synthetic strategies have
been developed for the efficient synthesis of polyketides. This paper first
introduces polyketides from different sources and classifications, then the
reconstruction of biosynthetic pathways is described using a “bottom-up”
synthetic biology approach. Through methods such as enhancing precursors,
relieving feedback inhibition, and dynamic regulation, the efficient production
of polyketides is achieved. Finally, the challenges faced by polyketides
research and future development directions are discussed.
Keywords:
Synthetic biology; Polyketides;
Polyketide synthases; Pathway refactoring; Yield
1. Introduction
In nature, natural products (also known as secondary metabolites, SMs) produced by plants, fungi, and bacteria are diverse and represent a vast treasure [1]. Polyketides, catalyzed by polyketide synthases (PKSs), are a significant component of these natural products. Their diverse chemical structures, including macrolides and aromatic polyketides, confer broad biological activities and application value [2,3]. In organisms, PKs can function as signaling molecules [4] and protect against external threats [5]. Melanin, a pigment found in mammals, plants, and microorganisms, has multiple biological functions such as UV radiation protection and free radical scavenging, enhancing the survival ability of organisms in harsh environments [6,7]. In the medical field, erythromycin, a macrolide antibiotic, is widely used clinically to treat Gram-positive bacterial infections, addressing health needs. Additionally, PKs demonstrate a range of biological activities, including antitumor (tetracycline), anticancer (doxorubicin), antiparasitic (avermectin, ivermectin), and immunosuppressive (rapamycin, FK506) properties [8,9,10] (Figure 1). In agriculture, spinosad, due to its low toxicity and highly efficient insecticidal activity, is used to control agricultural pests and ensure food security, earning the U.S. “Presidential Green Chemistry Challenge Award” in 1999. In the food industry, natamycin is widely utilized as a food preservative to inhibit fungal growth [11].
PKs have significant potential for applications and research. However, the indiscriminate use of antibiotics and pesticides has led to the emergence of multidrug-resistant (MDR) microbes and pests, it is necessary to develop new compounds through structural modification or genomic mining to meet practical needs. Additionally, the low yield of polyketides limits their large-scale production and application for several reasons: (1) Primary metabolites in cells maintain basic metabolic activities and growth, while SMs often primarily fulfill specific physiological functions and are typically non-essential [12]; (2) As significant microbial sources of polyketides, Streptomyces and fungi harbor multiple SMs biosynthesis gene clusters (BGCs) in their genomes, leading to low precursor conversion flux and by-product formation [13,14,15]; (3) Polyketides have complex structures, and their biosynthesis pathways involve multiple enzymes, with core enzymes (PKS) having multiple modules and domains, making genetic modification inefficient and challenging to synthesize using conventional chemical methods.
Advances in synthetic biology and metabolic engineering provide effective strategies for the development and yield enhancement of natural products. This article provides a systematic summary of polyketides, highlights the challenges of the biosynthesis of polyketides and the corresponding solution strategies. Firstly, the sources and classifications of polyketides are introduced. Then, the importance of modifying key enzymes and designing synthetic biology parts to refactor the biosynthetic pathway of polyketides is highlighted. Finally, it summarizes strategies for the efficient synthesis of polyketides and discusses future development directions and approaches. This paper proposes the “point (enzymes)—line (synthetic pathway)—plane (metabolic pathway)” strategy based on synthetic biology, which provides a theoretical foundation and technical support for the accelerated development and efficient production of polyketides.
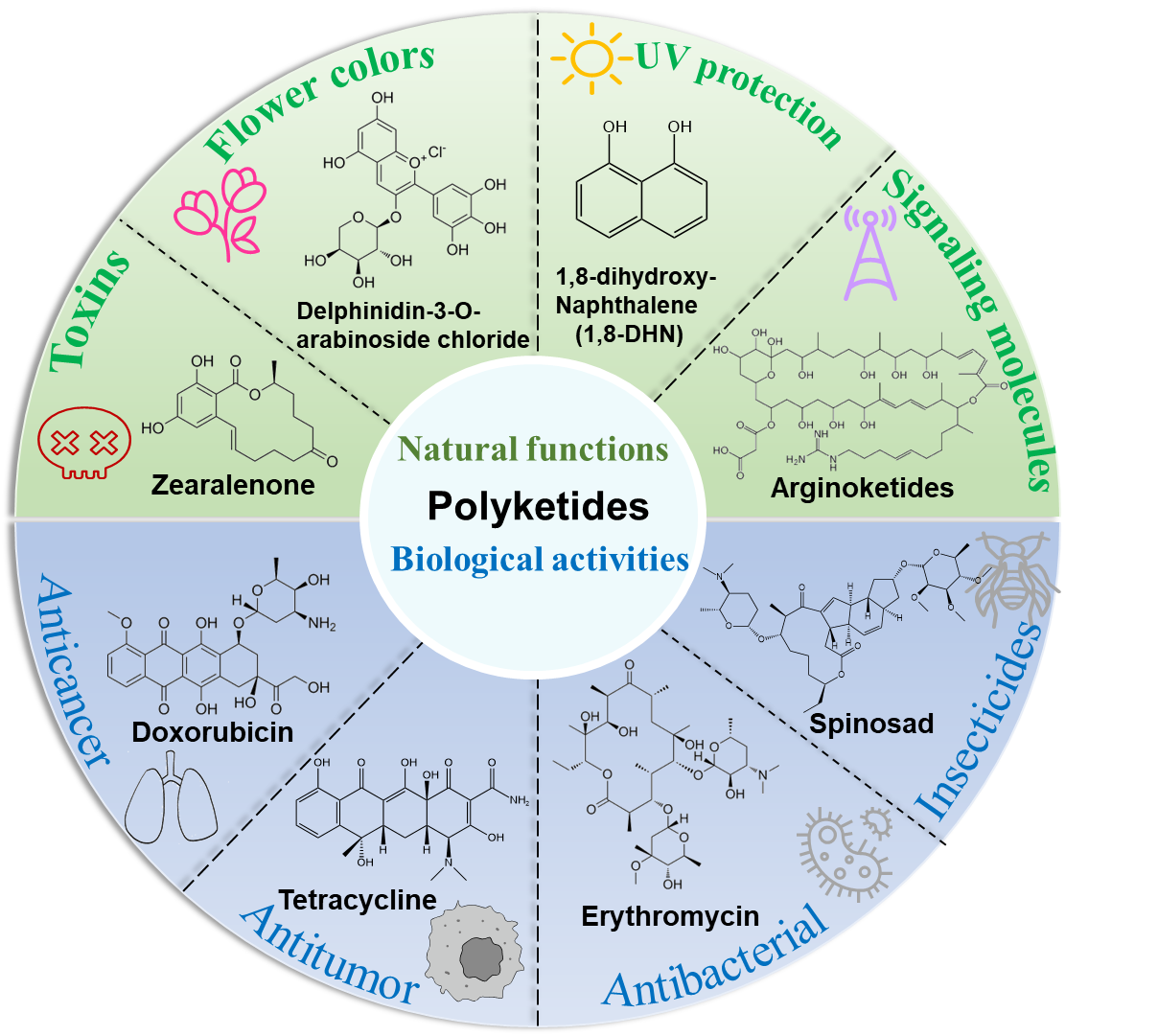
Figure 1. Diverse biological activities of polyketides. Delphinidin-3-O-arabinoside chloride is a PKS-derived product, which is formed through further modifications by using the PKS-mediated synthesis of the key intermediate chalcone; 1,8-DHN is the immediate precursor of melanin synthesis, which synthesized by PKS utilizing acetyl-CoA or malonyl-CoA.
2. Sources and Classification of Polyketides
2.1. Sources of Polyketides
Currently, the polyketides in widespread use generally come from two sources: (1) natural compounds isolated and identified from nature; (2) novel compounds generated by modifying the structures of natural compounds through genetic engineering or chemical derivatization to enhance biological activities or to develop new compounds.
2.1.1. Natural Polyketides
Natural polyketides are produced directly by bacteria, fungi, and plants. Actinorhodin, produced by Streptomyces coelicolor, is not only utilized as both an antibiotic and a natural blue pigment. It is one of the most effective model compounds for studying the regulatory mechanisms of secondary metabolism and gene editing methods in Streptomyces. For example, knocking out a gene in its BGC can be determined by the disappearance of the blue color to determine the effect of gene editing [16]. Other polyketides with antibiotic activity are often produced by Streptomyces and fungi (Table 1). Many plant-derived PKs also have natural pigment activities [17] and possess various physiological activities, including anti-inflammatory, antioxidant, obesity control, and immune system enhancement [18]. For example, Curcumin, a rare diketone compound mainly found in the rhizome of Curcuma longa. Due to its yellow color and special antioxidant structure, it is commonly used as a food coloring agent, preservative, and it can also be used to treat diabetes [19].
However, natural polyketides are also harmful to human health, including zearalenone [20] and fumonisin B1 [21]. Aflatoxins are a class of toxic secondary metabolites produced by Aspergillus flavus and Aspergillus parasiticus, including toxins and toxic alcohols such as B1, B2, G1, G2, and M1, of which B1 is the most toxic [22]. These compounds exhibit significant hepatotoxicity, hepatocarcinogenicity and teratogenicity.
2.1.2. Synthetic Polyketides
In response to multi-drug resistance, there is an urgent need to develop new drugs with novel targets or high activity. Direct screening from nature is time-consuming and labor-intensive, therefore, a current research focus is structural modification of existing compounds to produce new derivatives or to improve their biological properties.
Avermectin is an insecticide derived from Streptomyces avermitilis, the reduction of the C22–C23 double bond of avermectin B1 by using Wilkinson’s homogenous hydrogenation catalyst (PH3P)3RhCl, resulting in the ivermectin with enhanced insecticidal activity (Figure 2). In 2015, Satoshi Omura and William C. Campbell were awarded the Nobel Prize in Physiology/Medicine, along with Youyou Tu who discovered artemisinin, for their work on avermectin and ivermectin. Currently, ivermectin and moxidectin show significant potential in the treatment of human parasitic infections [37]. Roxithromycin and telithromycin are both derived from chemical modifications of erythromycin A, and exhibit improved medical efficacy [38]. Due to the highly complex structure of polyketides and the numerous intermediates involved, using chemical methods for synthesis or structural modification presents challenges such as multiple steps and low yields.
Recently, Dong et al. synthesized an analogue of the core TDO (2H-tetrahydro-4,6-dioxo-1,2-oxazine) heterocycle of alchivemycin A using chemical methods for the first time, then applied continuous oxidative modifications using enzymatic methods to obtain the final synthetic product. However, reconstructing the complex TDO heterocycle structure remains challenging [39]. Therefore, a better approach involves starting from the biosynthesis of the compounds themselves, utilizing the unique properties of microorganisms, and using molecular biology techniques to specifically manipulate key genes to synthesize specific natural products and their analogs. Koch et al. expanded the substrate utilization range by merely replacing key domains of PKS to obtain a series of pikromycin derivatives [40] that demonstrated the advantages of biosynthesis for expanding the structural diversity of natural products.
2.2. Classification of Polyketides
PKSs are key enzymes for the synthesis of polyketides. Based on their structure and synthesis mechanisms, PKSs can be classified into three major types: Type I PKSs (mainly synthesizing macrolides), Type II PKSs (synthesizing aromatic polyketides), and Type III PKSs (synthesizing flavonoids).
2.2.1. Type I PKSs
Type I PKSs are primarily found in bacteria and fungi, and exist as multifunctional enzymes in modular form. Each module contains non-redundant catalytic domains, with each domain participating in only one step of the polyketide chain elongation, to linearly complete the initiation and elongation of the polyketide synthesis (Table 2) [41,42,43,44]. The assembled polyketide chain is released via the thioesterase (TE) domain through cyclization or hydrolysis and subsequently modified by glycosyltransferases (GT), methyltransferases (MT), as well as oxidases such as monooxygenases (MO), cytochrome P450 (CYP450), and oxidoreductases (OR), to form polyketides with more complex structures and specific biological activities. The most representative compounds include erythromycin, rapamycin, and avermectin.
Butenyl-spinosyn studied in our laboratory is a green biopesticide produced by Saccharopolyspora pogona and also a typical type I polyketide. Its biosynthetic pathway includes 23 genes, of which busA~E encoding Type I PKSs are present in clusters on the genome (Figure 3a). The entire process involves the synthesis and cyclization of the polyketide chain, synthesis and attachment of side-chain sugars (L-rhamnose and D-forosamine) [45,46] (Figure 3b). Figure 3 depicts the biosynthetic process of Type I polyketides, with butenyl-spinosyn as an example.
The modular type I PKSs (mPKSs) described above are commonly found in bacteria, whereas iterative Type I PKSs (iPKSs) are central to biosynthesis in most fungi, iPKSs contain only one module that is reused, and the structural domains within the module are the same to those of the mPKSs. [47]. Based on the reduction degree of polyketide intermediates, iPKSs can be further classified into non-reducing (NR)-PKSs, partially reducing (PR)-PKSs, and highly reducing (HR)-PKSs [48,49]. The specific biosynthesis process is similar as mPKSs, with the distinction that intermediates in iPKSs are transferred back to the upstream KS domain until the final product is formed, while in mPKSs, intermediates continue to the downstream KS domain [50,51]. Another typical Type I PKS is trans-AT PKSs, which lacks the AT domain. Instead, AT activity is provided by one or more separate proteins within or outside the polyketide-BGC [52].
It is widely accepted that the domain composition within Type I PKS modules includes KS + AT (+DH + ER + KR) + ACP (+TE). However, Abe et al. studies on PKSs in aminopolyols revealed that the KS domain is located downstream of the processing enzymes in each module, redefining the PKS module as AT (+DH + ER + KR) + ACP + KS [53,54], this model is also applicable to trans-AT PKSs [55]. However, further research is needed for engineering applications.
2.2.2. Type II PKSs
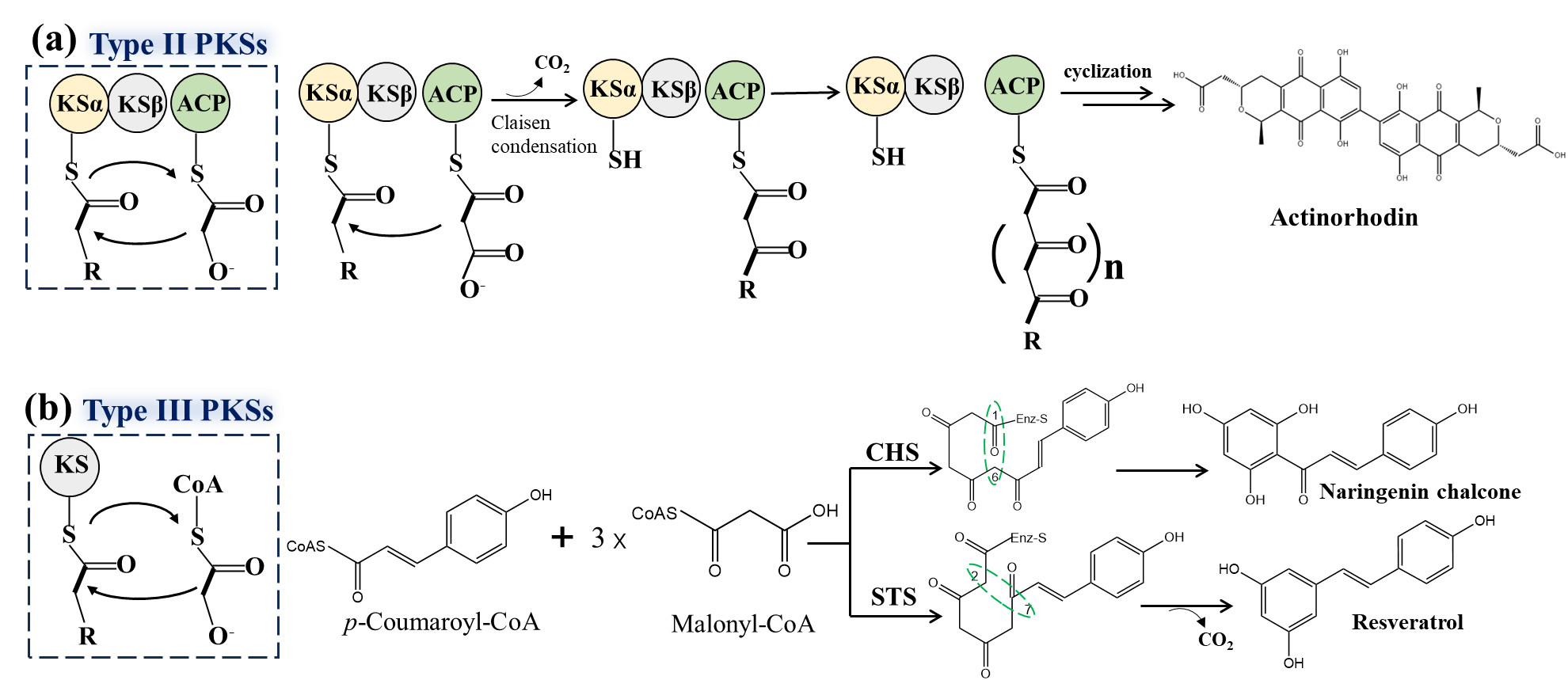
Type II PKSs, typically found in bacteria, are multi-enzyme complexes composed of discrete single-domain enzymes, which repeatedly use their functional domains to catalyze the same reactions at different stages. The minimal PKS consists of KSα, KSβ, and ACP [56], where KSβ controls the length of the polyketide chain and is also known as the chain length factor (CLF). The KSα-KSβ heterodimer (KS-CLF) repeatedly catalyzes multiple Claisen condensation reactions, which produces a poly-β-ketoacyl thioester intermediate bound to ACP. Under the action of KR, cyclase (CYC, mediating C9-C14 cyclization), and aromatase (ARO, mediating C7-C12 cyclization), the full-length poly-β-ketone intermediates are formed and then converted into aromatic scaffolds (Figure 4a). Finally, through the action of tailoring enzymes, these intermediates are transformed into aromatic polyketides, such as anthracycline and tetracycline [57,58].
2.2.3. Type III PKSs
Type III PKSs are primarily found in plants and are composed of 40–45 kDa KS homodimers. Their structure is simpler and they do not require ACP involvement. Each subunit possesses multiple activities, iteratively catalyzing initiation, elongation, and cyclization at the same active site. Type III PKSs mainly catalyze the biosynthesis of mono- and bicyclic aromatic polyketides [59,60,61]. The KS domain typically maintains the “Cys-His-Asn” catalytic triad, using CoA as the starter substrate, binding to the active site Cys, and using malonyl-CoA as the extender unit to perform repeated decarboxylative condensations to produce poly-β-ketone intermediates. These intermediates are then cyclized or released as linear products and further modified to synthesize polyketides with diverse biological activities.
Chalcone synthase (CHS) in plants is a representative Type III PKS. As a key enzyme in the biosynthetic pathway of flavonoids, CHS catalyzes the sequential condensation of three acetate units from malonyl-CoA to p-coumaroyl-CoA, forming a tetraketide intermediate. This intermediate undergoes Claisen-type cyclization to produce the aromatic tetraketide naringenin chalcone, which is further converted into flavonoids such as naringenin and icariin. Stilbene synthase (STS) functions similarly to CHS, but in the cyclization of the tetraketide intermediate, it catalyzes an intramolecular C2→C7 aldol-type condensation to form resveratrol (Figure 4b) [59].
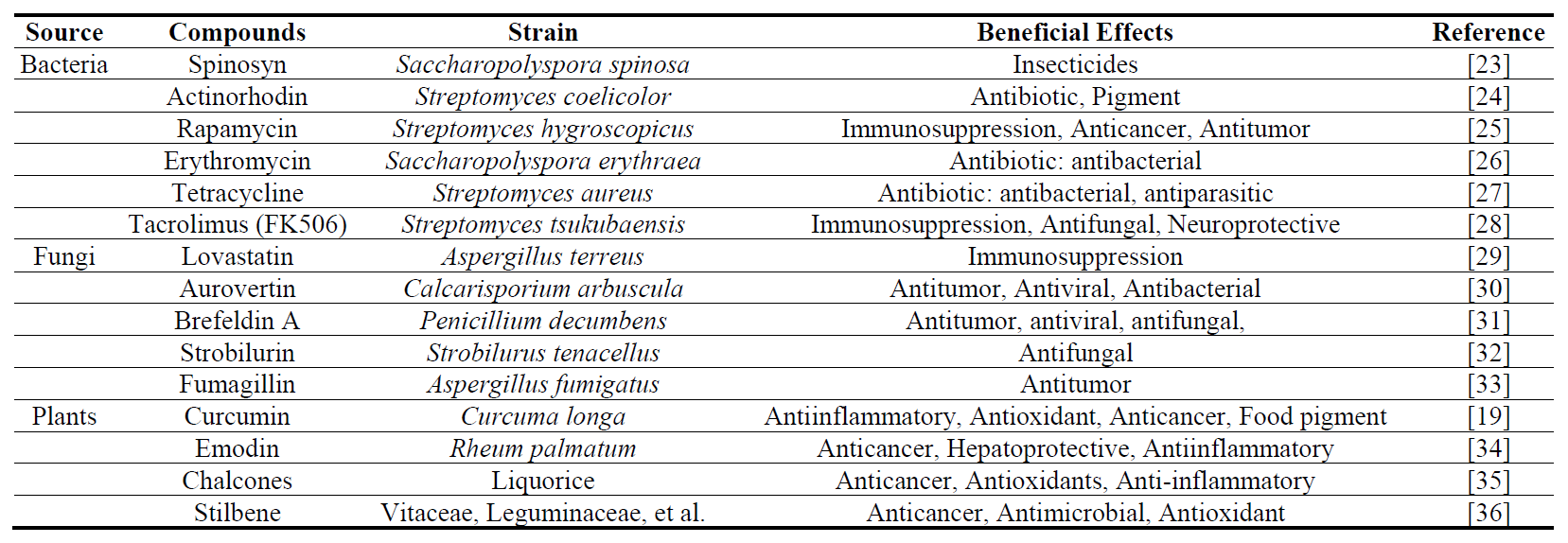
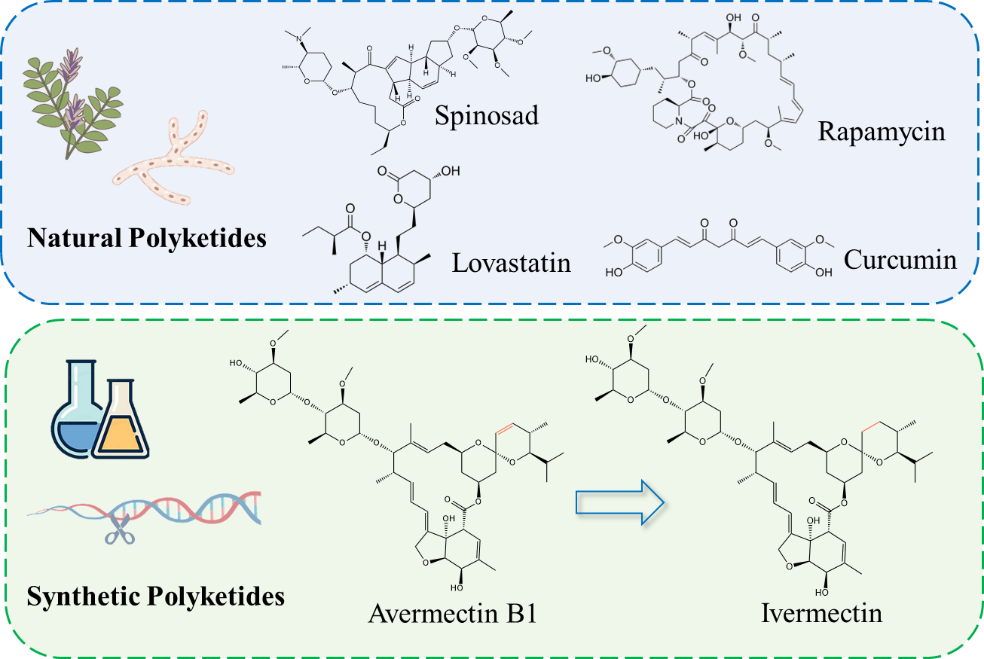
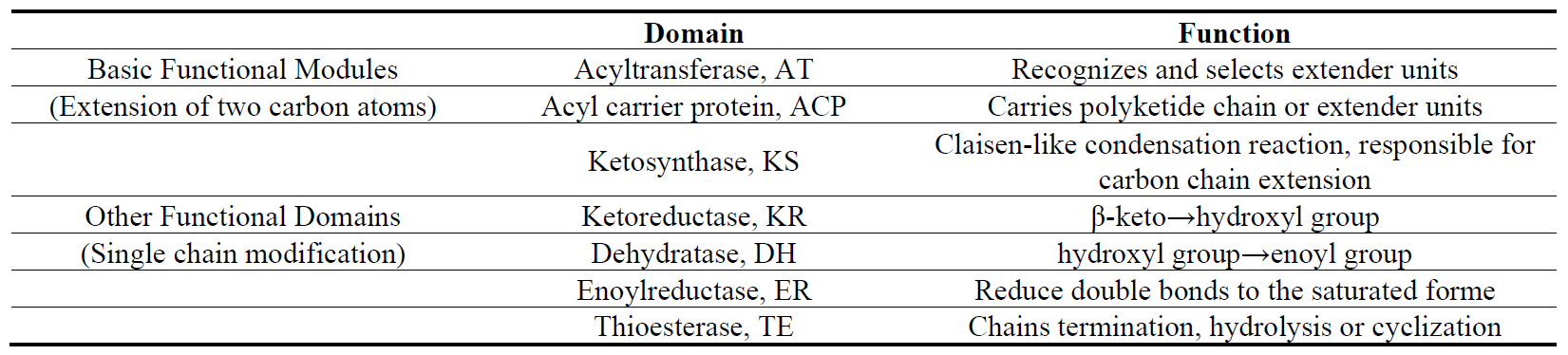
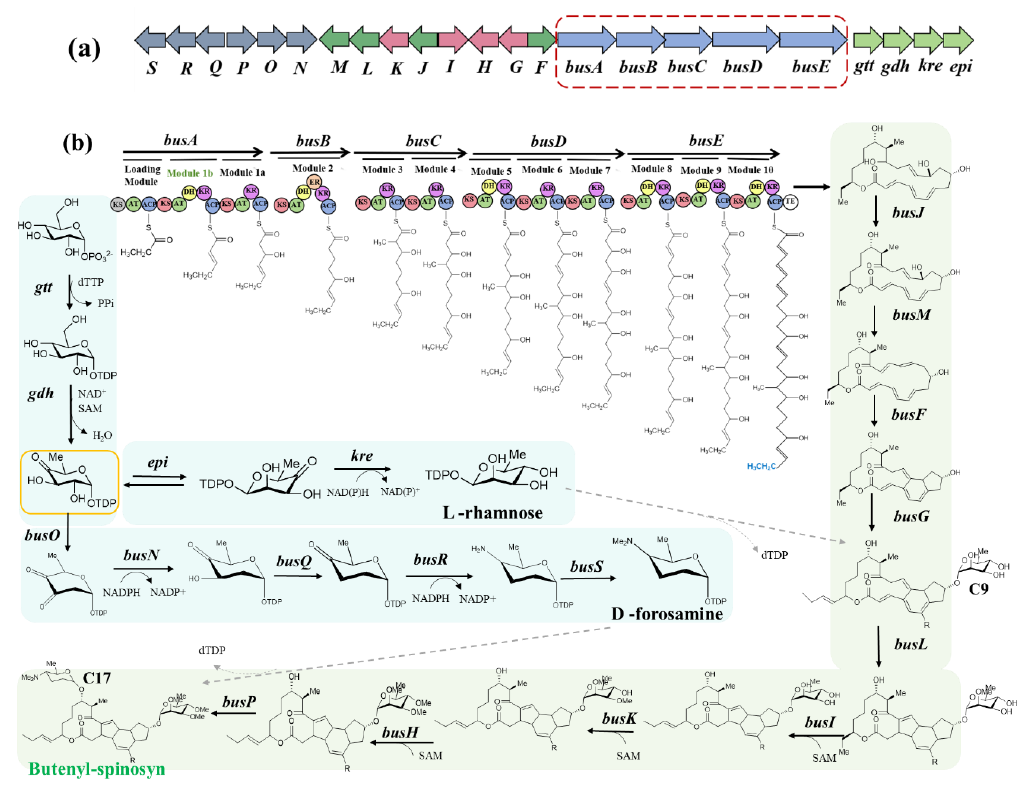
Figure 3. Biosynthesis of butenyl-spinosyn: (<b>a</b>) Butenyl-spinosyn BGC; (<b>b</b>) Biosynthetic pathway of butenyl-spinosyn. <i>gtt</i>: glucose-1-phosphate nucleotidyltransferase; <i>gdh</i>: glucose dehydrogenase; <i>epi</i>: 3′,5′-epimerase; <i>kre</i>: 4′-ketoreductase; <i>busO</i>: 2,3-dehydrogenase; <i>busN</i>: 3-ketoreductase; <i>busQ</i>: 3,4-dehydrogenase; <i>busR</i>: aminotransferase; <i>busS</i>: methyltransferases; <i>busG</i>: rhamnosyltransferase; busH: 2′-O-methyltransferases; <i>busI</i>: 3′-O-methyltransferases; <i>busK</i>: 4′-O-methyltransferases; <i>busJ</i>: dehydrogenase (catalyses C15 hydroxyl to carbonyl); <i>busM</i>: dehydratase(catalyses the formation of double bonds from C11-C12); <i>busF</i>: catalyses [4+2] cycloaddition bonding between C4-C12 and C7-C11; <i>busL</i>: catalyses Rauhut-Currier bonding of C3-C14; <i>busP</i>: forosaminyl transferase.
3. Synthetic Design of New Structures and High Production of Polyketides
3.1. Enzyme Engineering for New Structures of Polyketides
The differences in catalytic mechanisms and substrate specificities of PKSs result in the diversity in polyketide chain synthesis. A series of post-modification reactions further increase the structural diversity of polyketides, making it possible to engineer both PKSs and post-modification enzymes to produce new non-natural polyketides.
3.1.1. PKSs
The number and function of PKS modules and domains determine substrate selection, the extent of reduction and the stereochemistry of the products in the polyketide synthesis pathway. The number of modules defines the size of the backbone, and the domain composition controls the degree of functionalization [62]. Therefore, PKS modification can often enhance the structural diversity and biological activity of polyketides. This is mainly achieved through site-directed mutagenesis, as well as the insertion and replacement of domains and modules (Figure 5, Table 3). Type I PKSs are the most typical and complex of the three PKSs, therefore, this section focuses on Type I PKSs.
Site-directed mutation has minimal impact on the overall protein structure, however, it requires specific conditions regarding the size of the side chain and its flexibility [63,64]. In the biosynthesis pathway of FK506, the AT4FkbB domain can use both allylmalonyl and emthylmalonyl to produce the target product FK506 and its analog FK520. Molecular dynamics simulations revealed that the V187K mutation enhances substrate specificity and reduces the production of the by-product FK520 [65]. This strategy is often combined with domain and module engineering.
The starter and extender units largely determine the structural diversity and chemical complexity of polyketides. For example, the AT domain is responsible for CoA-based starter and extender units, and shows greater substrate promiscuity than other domains. Sheehan et al. replaced the loading module of the spinosad PKS with the loading module from the erythromycin PKS and added a range of carboxylic acids exogenously, resulting in 16 new spinosad derivatives. Among them, 21-cyclobutyl-spinosyn A along with its semisynthetic 5,6-dihydro derivative exhibited improved insecticidal activity [66]. Sirirungruang et al. introduced an F190V mutation into the wild-type trans-AT from the disorazole biosynthetic pathway (DszAT) and loaded it onto a specific PKS module in which the cis-AT was inactivated, and selectively introduced fluorine into the polyketide backbone and producing two fluorinated 6dEB analogs, 2-fluoro-2-desmethyl 6dEB and 4-fluoro-4-desmethyl 6dEB [67]. Domain engineering strategies are also applied to KR (responsible for the formation of stereocenters in polyketides) [68], DH [69], and TE in the unloading modules [70], although research in these areas remains relatively limited.
Another PKS engineering strategy for producing new compounds involves modifying domains or modules (including deletion, replacement, and addition), which directly influences product synthesis. Using RedEx technology, the integration of five domains (KS 1b, AT 1b, DH 1b, KR 1b, and ACP 1b) from module 1b of butenyl-spinosyn into the spinosad synthesis pathway between the KS and AT domains of module 1 successfully achieved butenyl group modification [71,72]. However, inserting heterologous domains often disrupts PKS stability, resulting in loss of activity and protein misfolding [73]. Appropriate linker regions and splice sites can retain protein interactions between donor and acceptor units during domain or module exchanges, reduce the impact of conformational changes during the polyketide chain elongation, and maintain overall PKS structure. Thus, splice sites are typically chosen within the AT and KS sequences or in linker regions upstream of ACP. However, the success rate of module exchanges is often low [74,75,76]. Yuzawa et al. systematically analyzed segments of AT domains and associated linkers in AT domain exchanges, identifying the module boundaries that maintain protein stability and enzyme activity when exchanging AT domains [77]. They subsequently developed a fluorescence-based solubility biosensor to rapidly detect AT-exchanged PKS hybrids with randomly assigned domain boundaries, thereby minimizing structural disruption [78].
Besides directly engineering domains, another approach involves starting with enzymes that exhibit high catalytic activity and broad substrate specificity. Zhang et al. discovered a unique β-subunit (Arm13) in acyl-CoA carboxylase (ACC) that combined with the α and ε subunits of propionyl-CoA carboxylase, forming a highly catalytic ACC. This ACC can recognize acyl-CoAs of different chain lengths and with various functional groups, efficiently converting them into corresponding alkylmalonyl-CoAs. By exogenously adding carboxylate precursors, a series of reactive functional groups such as alkenyl, alkynyl, and phenyl were introduced at the C6 position of the armeniaspirol backbone [79]. Zheng et al. found that the naturally lacking Lys-A10 residue in the acyl-CoA synthetases (ACSs) UkaQ in the UK-2A biosynthetic pathway enabled the synthesis of diverse acyl-CoAs. These, combined with permissive CCR and ACC, produced various malonyl-CoA extender units, resulting in the formation of new antimycin analogs [80]. This demonstrated the critical role of the catalytic activity of ACCase and ACSs in synthesizing polyketide extender units and new compounds.
3.1.2. Post-PKS Enzymes
After the synthesis of the polyketide chain, a series of post-synthetic modification reactions (such as methylation, glycosylation and acylation) are required to form the final polyketides. Modifying the post-PKS enzymes or related genes can alter the biological activity and structural diversity of end products.
The types of sugar moieties attached to the polyketide backbone can affect the biological activity, stability, and solubility of the compounds. Rhamnose not only is involved in the synthesis of spinosyn but also serves as a component in the synthesis of cell wall lipopolysaccharides. When rhamnose biosynthesis genes from S. spinosa are absent, different Streptomyces species cannot heterologously produce spinosyn [95]. Amphotericin B without mycosamine cannot trigger fungal membrane permeability, resulting in the loss of antifungal activity and toxicity [96]. Engineering natural product GTs can modify donor sugar, acceptor substrate, and acceptor position specificities to obtain natural products with unnatural glycosylation patterns [97,98,99]. For example, by replacing key amino termini of the C-glycosyltransferase encoded by the urdGT gene broadens and alters substrate specificity during the synthesis of urdamycin, resulting in the addition of glycosyl groups to unnatural substrates and the production of 28 new active urdamycins [100,101]. By deleting the genes responsible for the biosynthesis and attachment of TDP-D-desosamine in S. venezuelae ATCC 15439 and overexpressing genes encoding deoxysugar biosynthesis and glycosylation, various macrolide glycosyl derivatives were obtained [102]. Song et al. knocked out the spnK gene responsible for rhamnose methylation in the spinosyn biosynthetic gene cluster, thereby achieving heterologous biosynthesis of spinosyn JL [71]. Hydroxylation or other modifications by CYP450 also have the potential to lead to structural diversity [103]. The substrate-flexible CYP450 (pikC) from the S. venezuelae HK954 mutant produced new hydroxylated analogs of oleandomycin [104].
Modifying key enzymes is an effective way to generate new compounds. With the explosive development of big data and AI, the development of polyketides is not limited to substances with known synthetic pathways. In 2022, to further understand the abundance and diversity of aromatic polyketides from bacteria, Chen et al. constructed a phylogenetic tree and discovered a correlation between CLF enzyme amino acid sequences and compound structures. Using CLF as a marker, they identified 3254 bacterial Type II PKS gene clusters from databases, creating the first global atlas of bacterial aromatic polyketides. This allowed for the prediction of compound types synthesized from CLF gene information. Consequently, researchers identified a novel aromatic polyketide, oryzanaphthopyran, from Streptacidiphilus, therefore, making a breakthrough in the synthesis of new aromatic polyketides from bacterial Type II PKSs [105]. Due to the colinearity of the PKSs, tools like Alphafold and RoseTTAFold can combine natural PKS sequences to reversibly design specific PKSs biosynthetic pathways, generating millions of organic molecules and enabling the retrosynthesis of polyketides [106].
3.2. Synthesis Pathway Optimisation Enhances Adaptation for Polyketides Production
Eriko Takano proposed several design principles for synthetic biology of secondary metabolites. First, the biosynthetic gene clusters of secondary metabolites are identified through genome analysis. Then, the genes in the biosynthetic pathway are modularly designed and assembled based on the existing component library. Next, these genes are expressed in suitable screening hosts, with compatibility analyses conducted at the transcriptional level (e.g., promoters) and translational level (e.g., RBS). Finally, the best combinations are transferred into production hosts for production [107] (Figure 6). Based on these principles, deep mining of efficient functional parts, reconstructing metabolic pathways, and optimizing the adaptation of components and chassis to finely regulate metabolic flux to achieve high yields of target compounds.
3.2.1. Optimization of Biological Parts
Using synthetic biology to assemble biological parts in an orderly manner allows for easier and faster acquisition of compounds with desired properties. However, many parts exhibit incompatibility in different host systems, significantly reducing the biosynthetic efficiency of polyketides. Utilizing well-characterized genetic control elements can regulate gene expression at the transcriptional and translational levels.
Promoter engineering is a primary strategy to enhance the yield of natural products by regulating the expression of key genes. Using the CRISPR/cas9 system to replace the native promoters of limiting genes in the erythromycin biosynthetic gene cluster with the heterologous weak promoter PermE∗_s23 increased erythromycin yield by 6.0-fold, while the insertion of the strong promoter PkasO did not affect erythromycin yield, indicating that the use of compatible promoters to regulate gene expression can maximize the yield of target products [108]. Wang et al. obtained a strong endogenous promoter 5063p based on transcriptome analysis, which increased the expression of geldanamycin PKS by 4-141 times and geldanamycin yield by 39% when replacing the native PKS promoter gdmA1p [109]. Wei et al. mined the promoters of transcription factors related to secondary metabolism in A. nidulans, and, combined with single-cell fluorescence detection using flow cytometry, they obtained a natural promoter library with relative expression levels up to 37 times higher. Among them, the expression levels of PzipA and PsltA were 2.9 and 1.5 times higher than the constitutive promoter PgpdA, respectively [110]. Combining random mutagenesis of promoters and high-throughput screening is an efficient method to obtain promoters with a wide range of strengths. Tu et al. focused on droplet microfluidic-based high-throughput screening technology in Streptomyces, rapidly constructing promoter mutant libraries for subsequent fine regulation of metabolic pathways [111,112]. Artificial neural networks and computer-aided design can rationally design promoter sequences with specific requirements, avoiding interference from host regulatory mechanisms. The AI-assisted promoter sequence optimization method DeepSEED developed by Zhang et al, successfully designed promoters from various sources and types [113]. Terminators have less impact than promoters and are less studied and applied in Streptomyces. Horbal et al. mined a series of terminators from databases, including the Mycobacteria-derived terminator ttsbiB, which has a read-through of less than 4% [114].
Regulation alone at the transcriptional level is insufficient to construct highly efficient gene expression systems and optimization of translation efficiency is also required [115]. Translational regulation is primarily determined by the ribosome binding site (RBS) and the 5′ untranslated region (5′-UTR). In Streptomyces, an inappropriate RBS can reduce gene expression efficiency to zero. Researchers developed an in vivo RBS-selector using gusA as a reporter system to rapidly select optimal RBS for target genes, thereby rationally controlling protein expression levels [114]. The selection of RBS and the 5′ UTR is often combined with promoters. For example, by randomizing the strongest RBS sequence in S. venezuelae, a series of synthetic RBS with varying strengths were obtained and combined with promoters, identifying the optimal promoter-RBS combination for gene expression. This approach replaced the indigenous promoter and RBS sequences, achieving activation of silent gene clusters at different levels and mass production of target compounds [116]. Yi et al. screened two promoters and four 5′-UTR sequences from multiomics data of S. coelicolor, whose combination of which resulted in protein expression levels ranging from 0.03 to 2.4 times that of the strong promoter ermE∗p combined with the Shine-Dalgarno sequence [117]. The 5′-UTR limits the direct application of σ70-dependent promoters in Streptomyces. Replacing E. coli-derived 5′-UTR with Streptomyces-derived 5′-UTR converts σ70-dependent promoters into σhrdB-dependent promoters that efficiently are expressed in Streptomyces. RBS libraries were also constructed to optimize hybrid-5′-UTR. This strategy expands the sources of Streptomyces promoters and facilitates the construction of Streptomyces cell factories [118].
By combining the advantages of transcriptional and translational parts to design and control key enzymes in biosynthetic pathways can greatly promote the production of secondary metabolites. By integrating the development of biological parts with omics technologies that can mine important biological features and explore complex relationships between features, and by using machine learning and deep learning algorithms for model prediction, the accuracy and controllability of these elements have been significantly improved [119] (Figure 7).
3.2.2. Reconstruction of Synthetic Pathway
Reconstructing biosynthetic pathways to improve the synthesis of secondary metabolites is one of the core challenges for synthetic biology. Promoter replacement of rate-limiting genes in biosynthetic pathways with compatible ones is an effective strategy for the efficient production of target products. Tylosin, a 16-membered macrolide antibiotic used in veterinary medicine, consists of four structurally similar components: tylosin A, tylosin B, tylosin C, and tylosin D. By overexpressing the rate-limiting enzyme genes tylF and tylI with the strong promoter stnYp from Streptomyces flocculus CGMCC4.1223, the yield of tylosin A was elevated to 10.30 g/L, which was 1.7-fold higher than the industrial strain Streptomyces fradiae, the purity and yield of tylosin A were both improved [120]. Using a genome-scale metabolic network model (GSMM), overexpression of the four predicted key targets with kasO∗p increased pikromycin production in S. venezuelae [121]. The same strategy has also been applied to spinosyn [122] and actinorhodin [123].
BGCs display highly modular organization, and biosynthetic pathways typically involve the co-expression of multiple genes. According to the functions of the synthesis genes and the characteristics of the intermediates, they can be rearranged into different modules for fine regulation and coordinated expression, enhancing compatibility from module to module and from module to host. Song et al. took advantage of ExoCET multi-fragment assembly technology, divided the 79 kb biosynthetic gene cluster of spinosyn into seven operons and strengthened them with strong constitutive promoters, achieving efficient expression of spinosyn in S. albus J1074, with a 328-fold increase in yield compared to the native gene cluster [124]. Jiang et al. constructed a 106-kb multioperon artificial gene cluster, including five operons involved in natural salinomycin synthesis and five fatty acid β-oxidation genes into a single operon, driven by strong constitutive promoters, achieving efficient heterologous expression [125]. Shao et al. divided the BGC of type I polyketide spectinabilin into promoter modules, gene modules, and helper modules based on the modular design principle in synthetic biology. For promoter modules, strong promoters recognizable by the expression host were used to initiate transcription of exogenous genes, ensuring the expression of exogenous gene clusters; the gene modules contained eight genes required for spectinabilin biosynthesis; and the helper modules introduced elements that maintain the replication and selection of exogenous DNA within the host. Finally, the re-engineered and synthetically obtained gene cluster was cloned downstream of the promoter in the promoter module and introduced into S. lividans, resulting in a spectinabilin yield of 100 μg/L [126].
3.2.3. Adaptation of Chassis Hosts
Many hosts that synthesize polyketides face challenges such as slow growth, unsuitability for laboratory cultivation, or difficulty in genetic manipulation; these factors lead to extremely low yields or failure to synthesize many valuable PKs. Understanding biosynthetic mechanisms is critical to the rational selection and utilization of heterologous hosts are key to expressing BGCs for secondary metabolites. Many efficient and practical tools used for cloning and assembling large size-BGCs have been developed to capture the PKs synthesis pathway for heterologous expression in different hosts, e.g., TAR-cloning [127], AssemblX [128], MoClo [129], etc. Common heterologous expression strains include Streptomyces coelicolor, Streptomyces lividans, Streptomyces albus, Streptomyces avermitilis [130,131] and Aspergillus oryzae [132].
E. coli and S. cerevisiae are the most commonly used cell factories in synthetic biology. Although E. coli itself has difficulty in efficiently translating and folding key synthetic enzymes (such as PKS and CYP450) and cannot synthesize some acyl-CoA compounds, rational design and modification of the synthetic pathway can still achieve high-efficiency expression [133,134]. Curcumin is synthesized by a six-enzyme reaction, which is typically heterologously expressed in E. coli. Kang et al. used the multiplex automatic genome engineering (MAGE) tool in E. coli to mutate the 5′-UTR of each of the six genes, optimizing the expression balance of each enzyme. This led to a variant (6M08rv) that improved the curcumin yield by 38.2-fold [135]. P450 EryF is a key enzyme in the synthesis of erythronolide B (EB). After identifying the optimal SaEryF for EB biosynthesis and designing the I379V mutant based on its crystal structure, the EB yield in E. coli reached 131 mg/L [136], significantly reducing the synthesis cycle.
S. cerevisiae has been used to produce various simple polyketides, such as resveratrol and naringenin, and is also commonly used for the expression of fungal-derived BGCs [137,138]. Zhao et al. used promoter replacement and enzyme fusion strategies in S. cerevisiae to achieve the synthesis of bikaverin, with a yield of 202.75 mg/L [139]. These results demonstrate the enormous potential of E. coli and S. cerevisiae in producing complex polyketides.
3.3. Efficient Strategies for Production of Polyketides
Polyketide synthesis often involves balancing primary and secondary metabolism, and the biosynthetic pathways and regulatory mechanisms are complex. Simple metabolic pathway modifications may not significantly increase yields and may disrupt the metabolic network balance, leading to reduced target product yield or the generation of by-products. Therefore, to maximize the yield of target natural products, a global approach is adopted, enhancing precursor supply, relieving feedback inhibition, and employing dynamic balancing strategies.
3.3.1. Enhancing Precursor Supply
Primary metabolism provides precursors for secondary metabolism. By optimizing pathways related to precursor synthesis and removing competitive pathways, metabolic flux can be directed towards polyketide synthesis (Figure 8a). Liu et al. used transcriptome analysis to knock out the PEP phosphonomutase gene, leading to pyruvate accumulation and consequently increasing the precursor supply for spinosyn biosynthesis, increasing spinosyn A production by 2 times [140]. Genome-scale metabolic models (GEM) can predict metabolic flux distributions related to secondary metabolite precursors under specific conditions [141]. Under the guidance of GEM , knocking out the pfk gene encoding 6-phosphofructokinase resulted in upregulated pentose phosphate pathway flux, increasing NADPH levels and enhancing rapamycin production [142]. Oviedomycin, an anticancer polyketide synthesized by Type II PKS, Gu et al. used GEM to predict and overexpress the precursor and cofactor genes associated with enhanced oviedomycin synthesis. They also reconstructed the gene cluster by replacing the natural promoters of the oviedomycin BGC, the oviedomycin yield increased to 670 mg/L [143].
Fu et al. conducted a metabolomic analysis on the high-yield spinosyn strain ADE-AP and found that the high expression of PKS genes and acyl-CoA synthesis genes led to significant precursor accumulation, highlighting the critical role of precursor supply in yield improvement [144]. Malonyl-CoA, a crucial precursor for polyketide synthesis, is formed by the oxidative decarboxylation of pyruvate catalyzed by pyruvate dehydrogenase (PDH) to produce acetyl-CoA, followed by acetyl-CoA carboxylation. The natural PDH-ACC pathway has issues related to low catalytic efficiency and poor carbon utilization. Li et al. developed a non-carboxylative malonyl-CoA (NCM) pathway with a C3 (pyruvate)-C3 (3-oxopropanoate)-C3 (malonyl-CoA) catalytic mode, avoiding carbon loss, energy consumption and interference with the natural metabolic network, achieving high-efficiency synthesis of malonyl-CoA and its derivatives in different hosts, ultimately increasing spinosyn yield to 4.6 g/L [145].
Advancements in omics technologies have facilitated the discovery and analysis of unknown BGCs in genomes, a streptomyces genome typically encodes 25–50 secondary metabolite BGCs. The knockout of non-essential gene clusters can simplify the metabolic background of the strain and prevent precursor diversion to competitive secondary metabolite pathways. The S. pogona genome contains 32 gene clusters, including six polyketide gene clusters. The knockout of clu13 (a flaviolin-like gene cluster) increased butenyl-spinosyn yield by 4.06-fold [146]. Using the Latour gene editing system to knock out a ~20 kb NRPS-T1 PKS gene cluster increased butenyl-spinosyn yield by 4.72-fold [147]. Efficient heterologous expression of naringenin in Streptomyces albidoflavus J1074 can also be achieved through the deletion of competitive gene clusters [148].
3.3.2. Relieving Feedback Inhibition
The excessive intracellular accumulation of polyketides can impose metabolic burdens on the host or cause feedback inhibition of their synthesis pathways, thereby limiting yield increases. Some genes within the BGC can encode efflux proteins that can expel secondary metabolites from the cell, alleviating cytotoxicity and feedback inhibition, thereby increasing target product yield (Figure 8b). For example, the oxytetracycline (OTC) cluster encodes the membrane-bound protein OtrB, responsible for OTC efflux. Together with OtrA, a ribosomal protection protein, and OtrC, an ABC exporter, expelling the accumulation of excessive oxytetracycline, protecting the host strain from OTC toxicity, thereby enhancing the resistance level of Streptomyces rimosus to oxytetracycline and increasing oxytetracycline yield [149]. Some products also serve as mechanisms of relieving feedback inhibition. When intracellular rifamycin B levels are low, the regulatory factor RifQ negatively regulates the efflux protein RifP. As rifamycin B accumulates to a certain threshold, RifQ dissociates from rifP, avoiding excessive rifamycin B toxicity to the host. Overexpression of rifP and deletion of RifQ increased the yield of rifamycin more than 2-fold [150].
ATP-binding cassette transporters (ABC transporters) are crucial components of the Streptomyces transport system [151]. Daunorubicin (DNR) and its hydroxylated derivative doxorubicin (DXR), Type II polyketides produced by Streptomyces peucetius, exert anticancer effects by inserting into DNA and inhibiting type II topoisomerase. When DNR and DXR accumulate intracellularly, they inhibit the expression of transcriptional activators while inhibiting type II topoisomerase activity, leading to feedback inhibition of their synthesis. The DrrAB proteins encoded by S. peucetius drrAB, belonging to the ABC transporter family, can expel DNR, DXR ,and their structural analogs extracellularly, thus improving strain tolerance and increasing DNR and DXR yields [152,153,154]. The AvtAB proteins encoded by the avermectin BGC perform similar functions, resulting in a 50% increase in total avermectin B1a yield [155]. More and more transport proteins are discovered to promote the effective efflux and further yield enhancement of polyketides. Chu et al. developed a rational transporter screening process combined with a tunable plug-and-play exporter (TuPPE) module, discovering three new ABC transporters that universally increase polyketide yields [156]. These studies demonstrate that enhancing the efflux of end products in synthetic pathways is an effective strategy for relieving product feedback inhibition, increasing polyketide synthesis yields, and enhancing host strain tolerance.
3.3.3. Dynamic Regulation
The synthesis of secondary metabolites occurs in two stages. During the primary metabolism phase, cells consume external nutrients for rapid growth and reproduction; when external nutrients become limited, cell growth ceases, and the cells enter the secondary metabolism phase, where they begin producing secondary metabolites. How to increase SMs production without affecting normal growth is an important question. Dynamic regulation plays a crucial role in finely controlling biosynthesis (Figure 8c). The key lies in discovering and applying regulatory elements that respond to metabolic products. Using signals such as light, metabolites, or chemical molecules to design biosensors, cells can maintain a good production state, thereby improving the production performance of chassis cells [157].
Triacylglycerols (TAGs) are key metabolites that link primary and secondary metabolism, and regulate the metabolic switch [12,158]. The degradation of TAGs not only provides precursors and reduces power for the synthesis of polyketide but also redirects more carbon flux toward polyketide synthesis through changes in reducing power levels. Building on this insight, researchers developed the dynamic degradation of TAG (ddTAG) engineering strategy, which selectively controls the timing and strength of TAG degradation, significantly increasing the yield of various polyketides. The yield of avermectin B1 reached 9.31 g/L [12]. Besides using key metabolites for dynamic regulation, quorum sensing (QS) systems independent of metabolic pathways can achieve a dynamic balance. For example, Tian et al. combined QS with CRISPRi driven by γ-butyrolactones (GBLs)-responsive promoter to construct a novel dynamic regulation system called EQCi. This system can simultaneously regulate multiple targets. When integrated into S. rapamycinicus, it increased rapamycin production by approximately 660% without affecting cell growth, effectively balancing the metabolic flux between primary metabolism (cell growth) and secondary metabolism (product production) [159].
Currently, reconstructing biosynthetic pathways for secondary metabolites and heterologous expression are common metabolic engineering approaches. Changes in metabolic flux during this process may adversely affect cell growth and production. Combining GSMM and flux balance analysis (FBA) with secondary metabolic pathways can accurately simulate and predict the production flux of secondary metabolites [160]. From these results, biosensors can be designed, making the dynamic regulation system more efficient.
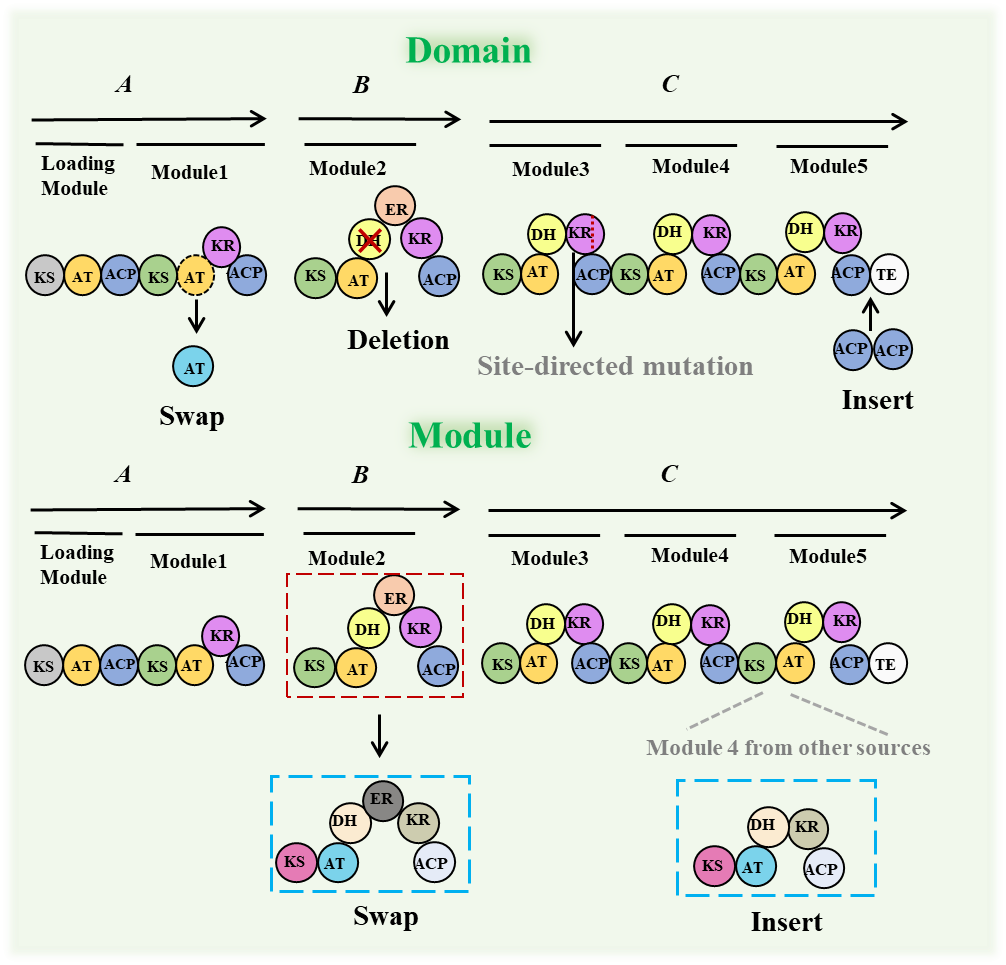
Figure 5. PKS modifications approach to obtain new polyketides. Deletion, swap and insertion of domains and modules and Site-directed mutation.
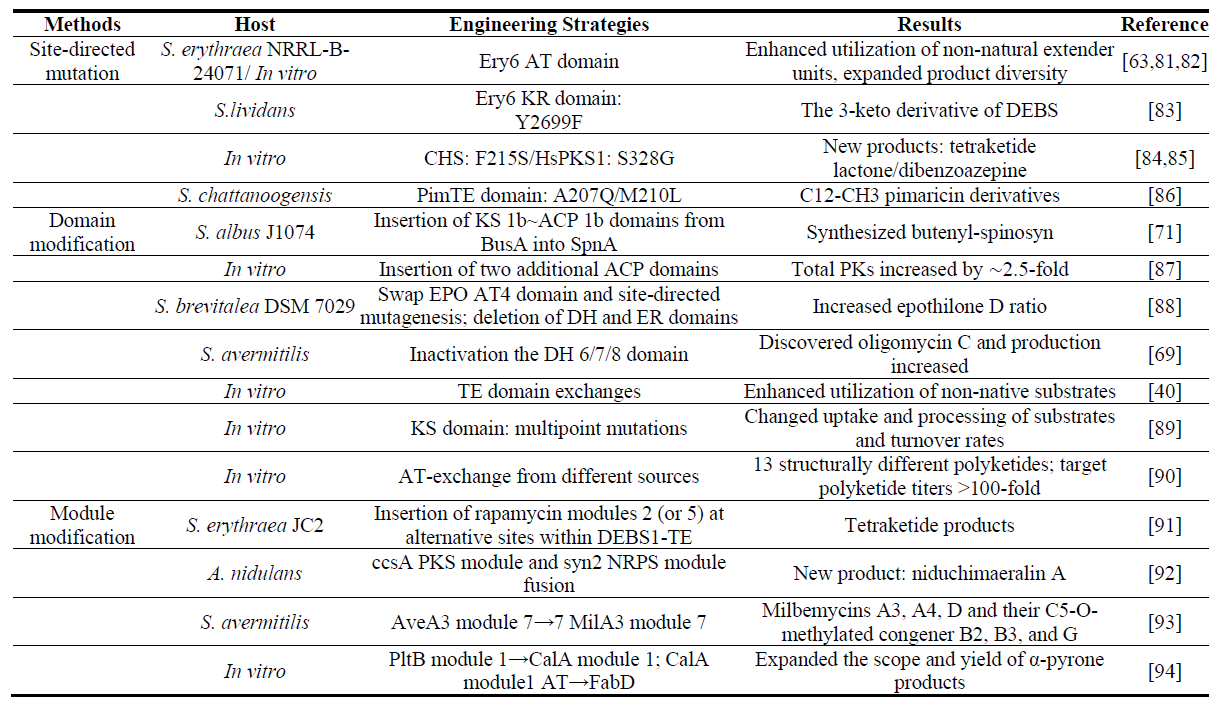
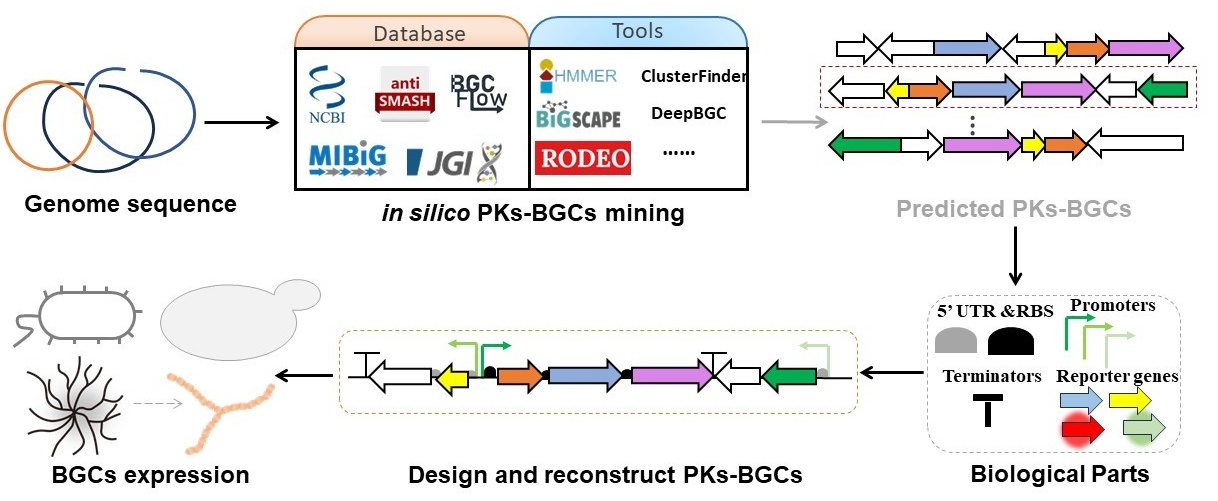
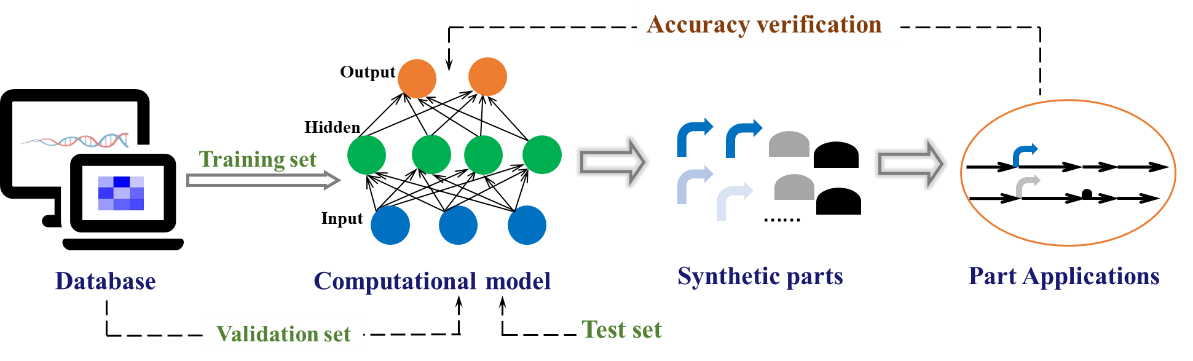
Figure 7. Artificial intelligence predicts biological parts. A portion of the existing sample databases is selected as the training set, and the computational model is selected to train the parameters, while the other portion of the sample databases is utilized as the validation set to refine and optimize the model parameters. After training is completed, the model is evaluated by using the test set. The model is then used to predict the biological parts that meet the requirements, and finally, the accuracy of the model is verified through actual experimental results.
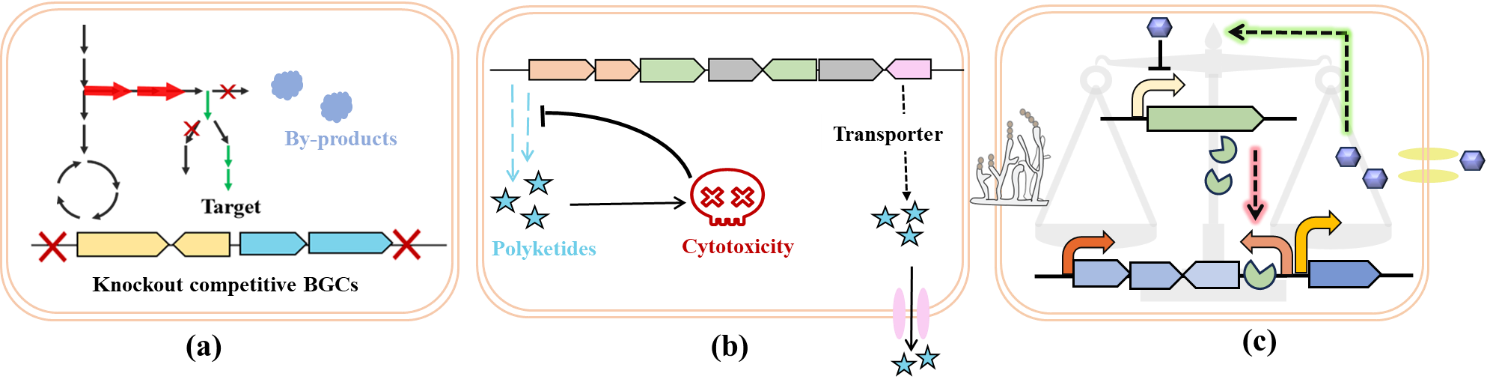
Figure 8. Efficient production strategies for polyketides. (<b>a</b>) Enhancing Precursor Supply; (<b>b</b>) Relieving feedback inhibition; (<b>c</b>) Dynamic Regulation.
4. Conclusions and Perspectives
Polyketides are a class of secondary metabolites with a wide range of biological activities, that have been developed into highly effective medical drugs, pesticides, and other products, with increasing market demand. However, most polyketides have naturally low yields, while plant-derived polyketides face challenges such as long growth cycles and low production efficiency. Additionally, the emergence of hard-to-treat diseases and pesticide-resistant pests necessitates accelerating the process of increasing target compound yields and discovering new compounds. A variety of strategies have been developed to address these issues. This paper focuses on the critical role of enzyme engineering in the development of new polyketides. Meanwhile, production can be enhanced by expanding the biological parts library, reconstructing metabolic pathways, and increasing precursor supply from multiple angles. However, excessive product accumulation can adversely affect cell growth and synthesis; therefore, global strategies such as strategies such as relieving feedback inhibition and balancing primary and secondary metabolism are proposed, dynamic regulation of key nodes in the synthesis process can ensure the efficient synthesis of target compounds.
With the rapid development and popularization of omics technologies, gene editing technologies, protein structure analysis and high-throughput sequencing technologies, an increasing number of polyketide biosynthetic pathways have been discovered and analyzed. Combined with rational bioinformatics design, engineering modifications can lead to more intelligent and diversified directions. For example, Wang et al. performed pan-genome analysis on Streptomyces, discovering the pqq gene co-evolving with polyketide BGCs, which enhanced the production of at least 16,385 metabolites and activated many unknown BGCs [161].
Despite significant progress in polyketide research, some areas remain under-researched. Currently, some special polyketides can be synthesized by wild strains only, and these are genetically more challenging to manipulate compared to traditional cell factories due to genomic characteristics and biochemical mechanisms. In the future, more efficient and universal genome editing technologies could be developed to obtain hosts with simpler genetic backgrounds, expand the range of model chassis and achieve efficient heterologous synthesis. The rapid development of protein analysis technology, especially the emergence of AlphaFold3, provides excellent tools for the accurate prediction of complex PKS structures, enabling “point-to-point” design and modification to further improve catalytic activity and efficiency. Melissa et al. used AlphaFold3 to accurately predict the transacylation site where polyketides can transfer from the acyl-ACP to the surface of a downstream KS domain [162]. On this basis, conducting protein dynamics studies to enhance interactions between domains and linkers, improving the efficiency of domain and module engineering. The synthesis of polyketides involves multiple complex and similar intermediate metabolites (e.g., the biosynthesis of butenyl- spinosyn, Figure 3b) and high-throughput screening and detection methods can be developed for real-time, quantitative detection, helping to better balance cell growth and product synthesis, achieving intelligent metabolic flux regulation. In conclusion, taking advantage of synthetic biology, combined with efficient genetic manipulation and metabolic engineering, it is expected that polyketides can be synthesized in a green and efficient way and promote large-scale production and application in different fields!
Author Contributions
Ethics Statement
Not applicable.
Informed Consent Statement
Funding
This research was funded by the National Key Research and Development Program of China (2023YFA0914700), the National Natural Science Foundation of China (22108153), the Foundation of Key Laboratory of Industrial Bi-ocatalysis Ministry of Education, Tsinghua University (No.2023003).
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
1.
Jakubczyk D, Dussart F. Selected Fungal Natural Products with Antimicrobial Properties. Molecules 2020, 25, 911. [Google Scholar]
2.
Mrudulakumari Vasudevan U, Lee EY. Flavonoids, Terpenoids, and Polyketide Antibiotics: Role of Glycosylation and Biocatalytic Tactics in Engineering Glycosylation. Biotechnol. Adv. 2020, 41, 107550. [Google Scholar]
3.
Grininger M. Enzymology of Assembly Line Synthesis by Modular Polyketide Synthases. Nat. Chem. Biol. 2023, 19, 401–415. [Google Scholar]
4.
Krespach MKC, Stroe MC, Netzker T, Rosin M, Zehner LM, Komor AJ, et al. Streptomyces Polyketides Mediate Bacteria–Fungi Interactions across Soil Environments. Nat. Microbiol. 2023, 8, 1348–1361. [Google Scholar]
5.
Liu L, Sasse C, Dirnberger B, Valerius O, Fekete-Szücs E, Harting R, et al. Secondary Metabolites of Hülle Cells Mediate Protection of Fungal Reproductive and Overwintering Structures against Fungivorous Animals. eLife 2021, 10, e68058. [Google Scholar]
6.
Singh S, Nimse SB, Mathew DE, Dhimmar A, Sahastrabudhe H, Gajjar A, et al. Microbial Melanin: Recent Advances in Biosynthesis, Extraction, Characterization, and Applications. Biotechnol. Adv. 2021, 53, 107773. [Google Scholar]
7.
Kordjazi T, Mariniello L, Giosafatto CVL, Porta R, Restaino OF. Streptomycetes as Microbial Cell Factories for the Biotechnological Production of Melanin. Int. J. Mol. Sci. 2024, 25, 3013. [Google Scholar]
8.
Weissman KJ, Leadlay PF. Combinatorial Biosynthesis of Reduced Polyketides. Nat. Rev. Microbiol. 2005, 3, 925–936. [Google Scholar]
9.
Benjamin D, Colombi M, Moroni C, Hall MN. Rapamycin Passes the Torch: A New Generation of mTOR Inhibitors. Nat. Rev. Drug Discov. 2011, 10, 868–880. [Google Scholar]
10.
Õmura S, Crump A. The Life and Times of Ivermectin—a Success Story. Nat. Rev. Microbiol. 2004, 2, 984–989. [Google Scholar]
11.
Meena M, Prajapati P, Ravichandran C, Sehrawat R. Natamycin: A Natural Preservative for Food Applications—a Review. Food Sci. Biotechnol. 2021, 30, 1481–1496. [Google Scholar]
12.
Wang W, Li S, Li Z, Zhang J, Fan K, Tan G, et al. Harnessing the Intracellular Triacylglycerols for Titer Improvement of Polyketides in Streptomyces. Nat. Biotechnol. 2020, 38, 76–83. [Google Scholar]
13.
van Bergeijk DA, Terlouw BR, Medema MH, van Wezel GP. Ecology and Genomics of Actinobacteria: New Concepts for Natural Product Discovery. Nat. Rev. Microbiol. 2020, 18, 546–558. [Google Scholar]
14.
Chen G, Wang M, Ni X, Xia H. Optimization of Tetramycin Production in Streptomyces Ahygroscopicus S91. J. Biol. Eng. 2021, 15, 16. [Google Scholar]
15.
Wang H, Liu Y, Cheng X, Zhang Y, Li S, Wang X, et al. Titer Improvement of Milbemycins via Coordinating Metabolic Competition and Transcriptional Co-activation Controlled by Streptomyces Antibiotic Regulatory Protein Family Regulator in Streptomyces Bingchenggensis. Biotechnol. Bioeng. 2022, 119, 1252–1263. [Google Scholar]
16.
Hua H-M, Xu J-F, Huang X-S, Zimin AA, Wang W-F, Lu Y-H. Low-Toxicity and High-Efficiency Streptomyces Genome Editing Tool Based on the Miniature Type V–F CRISPR/Cas Nuclease AsCas12f1. J. Agric. Food Chem. 2024, 72, 5358–5367. [Google Scholar]
17.
Iwashina T. Contribution to Flower Colors of Flavonoids Including Anthocyanins: A Review. Nat. Prod. Commun. 2015, 10, 529–544. [Google Scholar]
18.
Sun J, Sun W, Zhang G, Lv B, Li C. High Efficient Production of Plant Flavonoids by Microbial Cell Factories: Challenges and Opportunities. Metab. Eng. 2022, 70, 143–154. [Google Scholar]
19.
Kotha RR, Luthria DL. Curcumin: Biological, Pharmaceutical, Nutraceutical, and Analytical Aspects. Molecules 2019, 24, 2930. [Google Scholar]
20.
Cai P, Liu S, Tu Y, Shan T. Toxicity, Biodegradation, and Nutritional Intervention Mechanism of Zearalenone. Sci. Total Environ. 2024, 911, 168648. [Google Scholar]
21.
Chen J, Wei Z, Wang Y, Long M, Wu W, Kuca K. Fumonisin B1: Mechanisms of Toxicity and Biological Detoxification Progress in Animals. Food Chem. Toxicol. 2021, 149, 111977. [Google Scholar]
22.
Agriopoulou S, Stamatelopoulou E, Varzakas T. Advances in Occurrence, Importance, and Mycotoxin Control Strategies: Prevention and Detoxification in Foods. Foods 2020, 9, 137. [Google Scholar]
23.
Huang K, Xia L, Zhang Y, Ding X, Zahn JA. Recent Advances in the Biochemistry of Spinosyns. Appl. Microbiol. Biotechnol. 2009, 82, 13–23. [Google Scholar]
24.
Mak S, Nodwell JR. Actinorhodin Is a Redox-Active Antibiotic with a Complex Mode of Action against Gram-Positive Cells. Mol. Microbiol. 2017, 106, 597–613. [Google Scholar]
25.
Mannick JB, Lamming DW. Targeting the Biology of Aging with mTOR Inhibitors. Nat. Aging. 2023, 3, 642–660. [Google Scholar]
26.
Jelić D, Antolović R. From Erythromycin to Azithromycin and New Potential Ribosome-Binding Antimicrobials. Antibiotics 2016, 5, 29. [Google Scholar]
27.
Chopra I, Roberts M. Tetracycline Antibiotics: Mode of Action, Applications, Molecular Biology, and Epidemiology of Bacterial Resistance. Microbiol. Mol. Biol. Rev. 2001, 65, 232–260. [Google Scholar]
28.
Ban YH, Park SR, Yoon YJ. The Biosynthetic Pathway of FK506 and Its Engineering: From Past Achievements to Future Prospects. J. Ind. Microbiol. Biotechnol. 2016, 43, 389–400. [Google Scholar]
29.
Wang J, Liang J, Chen L, Zhang W, Kong L, Peng C, et al. Structural Basis for the Biosynthesis of Lovastatin. Nat. Commun. 2021, 12, 867. [Google Scholar]
30.
Cheng J-T, Yu J-H, Sun C-F, Cao F, Ying Y-M, Zhan Z-J, et al. A Cell Factory of a Fungicolous Fungus Calcarisporium Arbuscula for Efficient Production of Natural Products. ACS Synth. Biol. 2021, 10, 698–706. [Google Scholar]
31.
Paek S-M. Recent Synthesis and Discovery of Brefeldin A Analogs. Mar. Drugs 2018, 16, 133. [Google Scholar]
32.
Skellam E. Biosynthesis of Fungal Polyketides by Collaborating and Trans -Acting Enzymes. Nat. Prod. Rep. 2022, 39, 754–783. [Google Scholar]
33.
Lefkove B, Govindarajan B, Arbiser JL. Fumagillin: An Anti-Infective as a Parent Molecule for Novel Angiogenesis Inhibitors. Expert Rev. Anti Infect. Ther. 2007, 5, 573–579. [Google Scholar]
34.
Dong X, Fu J, Yin X, Cao S, Li X, Lin L, et al. Emodin: A Review of Its Pharmacology, Toxicity and Pharmacokinetics. Phytother. Res. 2016, 30, 1207–1218. [Google Scholar]
35.
Rammohan A, Reddy JS, Sravya G, Rao CN, Zyryanov GV. Chalcone Synthesis, Properties and Medicinal Applications: A Review. Environ. Chem. Lett. 2020, 18, 433–458. [Google Scholar]
36.
Teka T, Zhang L, Ge X, Li Y, Han L, Yan X. Stilbenes: Source Plants, Chemistry, Biosynthesis, Pharmacology, Application and Problems Related to Their Clinical Application-A Comprehensive Review. Phytochemistry 2022, 197, 113128. [Google Scholar]
37.
Sprecher VP, Hofmann D, Savathdy V, Xayavong P, Norkhankhame C, Huy R, et al. Efficacy and Safety of Moxidectin Compared with Ivermectin against Strongyloides Stercoralis Infection in Adults in Laos and Cambodia: A Randomised, Double-Blind, Non-Inferiority, Phase 2b/3 Trial. Lancet Infect. Dis. 2024, 24, 196–205. [Google Scholar]
38.
Chen D, Wu J, Liu W. Biosynthesis-based production improvement and structure modification of erythromycin A. Chin. J. Biotech. 2015, 31, 939–954. [Google Scholar]
39.
Dong H, Guo N, Hu D, Hong B, Liao D, Zhu HJ, et al. Chemoenzymatic Total Synthesis of Alchivemycin A. Nat. Synth. 2024, 1–10.
40.
Koch AA, Schmidt JJ, Lowell AN, Hansen DA, Coburn KM, Chemler JA, et al. Probing Selectivity and Creating Structural Diversity Through Hybrid Polyketide Synthases. Angew. Chem. Int. Ed. 2020, 59, 13575–13580. [Google Scholar]
41.
Dutta S, Whicher JR, Hansen DA, Hale WA, Chemler JA, Congdon GR, et al. Structure of a Modular Polyketide Synthase. Nature 2014, 510, 512–517. [Google Scholar]
42.
Barajas JF, Blake-Hedges JM, Bailey CB, Curran S, Keasling JD. Engineered Polyketides: Synergy between Protein and Host Level Engineering. Synth. Syst. Biotechnol. 2017, 2, 147–166. [Google Scholar]
43.
Weissman KJ. Genetic Engineering of Modular PKSs: From Combinatorial Biosynthesis to Synthetic Biology. Nat. Prod. Rep. 2016, 33, 203–230. [Google Scholar]
44.
Gayen AK, Nichols L, Williams GJ. An Artificial Pathway for Polyketide Biosynthesis. Nat. Catal. 2020, 3, 536–538. [Google Scholar]
45.
Shou J, Qiu L. A New Type of Biological Pesticide--Butenyl-spinosyns. Agrochemicals 2011, 50, 239–243+272. [Google Scholar]
46.
Guo C, Guo W, Liu Y, Wang C. Complete Genome Sequence of Butenyl-Spinosyn-Producing Saccharopolyspora Strain ASAGF58. Ann. Microbiol. 2020, 70, 46. [Google Scholar]
47.
Chooi Y-H, Tang Y. Navigating the Fungal Polyketide Chemical Space: From Genes to Molecules. J. Org. Chem. 2012, 77, 9933–9953. [Google Scholar]
48.
Hertweck C. The Biosynthetic Logic of Polyketide Diversity. Angew. Chem. Int. Ed. 2009, 48, 4688–4716. [Google Scholar]
49.
Cox RJ. Curiouser and Curiouser: Progress in Understanding the Programming of Iterative Highly-Reducing Polyketide Synthases. Nat. Prod. Rep. 2023, 40, 9–27. [Google Scholar]
50.
Wang B, Guo F, Huang C, Zhao H. Unraveling the Iterative Type I Polyketide Synthases Hidden in Streptomyces. Proc. Natl. Acad. Sci. USA 2020, 117, 8449–8454. [Google Scholar]
51.
Wang J, Deng Z, Liang J, Wang Z. Structural Enzymology of Iterative Type I Polyketide Synthases: Various Routes to Catalytic Programming. Nat. Prod. Rep. 2023, 40, 1498–1520. [Google Scholar]
52.
Chen H, Bian Z, Ravichandran V, Li R, Sun Y, Huo L, et al. Biosynthesis of Polyketides by Trans-AT Polyketide Synthases in Burkholderiales. Crit. Rev. Microbiol. 2019, 45, 162–181. [Google Scholar]
53.
Zhang L, Hashimoto T, Qin B, Hashimoto J, Kozone I, Kawahara T, et al. Characterization of Giant Modular PKSs Provides Insight into Genetic Mechanism for Structural Diversification of Aminopolyol Polyketides. Angew. Chem. Int. Ed. 2017, 56, 1740–1745. [Google Scholar]
54.
Keatinge-Clay AT. Polyketide Synthase Modules Redefined. Angew. Chem. Int. Ed. 2017, 56, 4658–4660. [Google Scholar]
55.
Nivina A, Yuet KP, Hsu J, Khosla C. Evolution and Diversity of Assembly-Line Polyketide Synthases: Focus Review. Chem. Rev. 2019, 119, 12524–12547. [Google Scholar]
56.
Bisang C, Long PF, Corte’s J, Westcott J, Crosby J, Matharu A-L, et al. A Chain Initiation Factor Common to Both Modular and Aromatic Polyketide Synthases. Nature 1999, 401, 502–505. [Google Scholar]
57.
Hertweck C, Luzhetskyy A, Rebets Y, Bechthold A. Type II Polyketide Synthases: Gaining a Deeper Insight into Enzymatic Teamwork. Nat. Prod. Rep. 2007, 24, 162–190. [Google Scholar]
58.
Bräuer A, Zhou Q, Grammbitter GLC, Schmalhofer M, Rühl M, Kaila VRI, et al. Structural Snapshots of the Minimal PKS System Responsible for Octaketide Biosynthesis. Nat. Chem. 2020, 12, 755–763. [Google Scholar]
59.
Yu D, Xu F, Zeng J, Zhan J. Type III Polyketide Synthases in Natural Product Biosynthesis. IUBMB Life 2012, 64, 285–295. [Google Scholar]
60.
Lim Y, Go M, Yew W. Exploiting the Biosynthetic Potential of Type III Polyketide Synthases. Molecules 2016, 21, 806. [Google Scholar]
61.
Katsuyama Y, Kita T, Funa N, Horinouchi S. Curcuminoid Biosynthesis by Two Type III Polyketide Synthases in the Herb Curcuma Longa. J. Biol. Chem. 2009, 284, 11160–11170. [Google Scholar]
62.
Peng H, Ishida K, Hertweck C. Loss of Single-Domain Function in a Modular Assembly Line Alters the Size and Shape of a Complex Polyketide. Angew. Chem. Int. Ed. 2019, 58, 18252–18256. [Google Scholar]
63.
Bravo-Rodriguez K, Klopries S, Koopmans KRM, Sundermann U, Yahiaoui S, Arens J, et al. Substrate Flexibility of a Mutated Acyltransferase Domain and Implications for Polyketide Biosynthesis. Chem. Biol. 2015, 22, 1425–1430. [Google Scholar]
64.
Sundermann U, Bravo-Rodriguez K, Klopries S, Kushnir S, Gomez H, Sanchez-Garcia E, et al. Enzyme-Directed Mutasynthesis: A Combined Experimental and Theoretical Approach to Substrate Recognition of a Polyketide Synthase. ACS Chem. Biol. 2013, 8, 443–450. [Google Scholar]
65.
Shen J-J, Chen F, Wang X-X, Liu X-F, Chen X-A, Mao X-M, et al. Substrate Specificity of Acyltransferase Domains for Efficient Transfer of Acyl Groups. Front. Microbiol. 2018, 9, 1840. [Google Scholar]
66.
Sheehan LS, Lill RE, Wilkinson B, Sheridan RM, Vousden WA, Kaja AL, et al. Engineering of the Spinosyn PKS: Directing Starter Unit Incorporation. J. Nat. Prod. 2006, 69, 1702–1710. [Google Scholar]
67.
Sirirungruang S, Ad O, Privalsky TM, Ramesh S, Sax JL, Dong H, et al. Engineering Site-Selective Incorporation of Fluorine into Polyketides. Nat. Chem. Biol. 2022, 18, 886–893. [Google Scholar]
68.
Schröder M, Roß T, Hemmerling F, Hahn F. Studying a Bottleneck of Multimodular Polyketide Synthase Processing: The Polyketide Structure-Dependent Performance of Ketoreductase Domains. ACS Chem. Biol. 2022, 17, 1030–1037. [Google Scholar]
69.
Wang J, Wang K, Deng Z, Zhong Z, Sun G, Mei Q, et al. Engineered Cytosine Base Editor Enabling Broad-Scope and High-Fidelity Gene Editing in Streptomyces. Nat. Commun. 2024, 15, 5687. [Google Scholar]
70.
Tripathi A, Choi S-S, Sherman DH, Kim E-S. Thioesterase Domain Swapping of a Linear Polyketide Tautomycetin with a Macrocyclic Polyketide Pikromycin in Streptomyces Sp. CK4412. J. Ind. Microbiol. Biotechnol. 2016, 43, 1189–1193. [Google Scholar]
71.
Song C, Luan J, Li R, Jiang C, Hou Y, Cui Q, et al. RedEx: A Method for Seamless DNA Insertion and Deletion in Large Multimodular Polyketide Synthase Gene Clusters. Nucleic Acids Res. 2020, 48, e130. [Google Scholar]
72.
Luan J, Song C, Liu Y, He R, Guo R, Cui Q, et al. Seamless Site-Directed Mutagenesis in Complex Cloned DNA Sequences Using the RedEx Method. Nat. Protoc. 2024, 1–29. doi: 10.1038/s41596-024-01016-9.
73.
Menzella HG, Reid R, Carney JR, Chandran SS, Reisinger SJ, Patel KG, et al. Combinatorial Polyketide Biosynthesis by de Novo Design and Rearrangement of Modular Polyketide Synthase Genes. Nat. Biotechnol. 2005, 23, 1171–1176. [Google Scholar]
74.
Wu N, Tsuji SY, Cane DE, Khosla C. Assessing the Balance between Protein−Protein Interactions and Enzyme−Substrate Interactions in the Channeling of Intermediates between Polyketide Synthase Modules. J. Am. Chem. Soc. 2001, 123, 6465–6474. [Google Scholar]
75.
Su L, Hôtel L, Paris C, Chepkirui C, Brachmann AO, Piel J, et al. Engineering the Stambomycin Modular Polyketide Synthase Yields 37-Membered Mini-Stambomycins. Nat. Commun. 2022, 13, 515. [Google Scholar]
76.
Zhang L, Hashimoto T, Qin B, Hashimoto J, Kozone I, Kawahara T, et al. Characterization of Giant Modular PKSs Provides Insight into Genetic Mechanism for Structural Diversification of Aminopolyol Polyketides. Angew. Chem. Int. Ed. 2017, 56, 1740–1745. [Google Scholar]
77.
Yuzawa S, Deng K, Wang G, Baidoo EEK, Northen TR, Adams PD, et al. Comprehensive in Vitro Analysis of Acyltransferase Domain Exchanges in Modular Polyketide Synthases and Its Application for Short-Chain Ketone Production. ACS Synth. Biol. 2017, 6, 139–147. [Google Scholar]
78.
Englund E, Schmidt M, Nava AA, Klass S, Keiser L, Dan Q, et al. Biosensor Guided Polyketide Synthases Engineering for Optimization of Domain Exchange Boundaries. Nat. Commun. 2023, 14, 4871. [Google Scholar]
79.
Zhang J, Zheng M, Yan J, Deng Z, Zhu D, Qu X. A Permissive Medium Chain Acyl-CoA Carboxylase Enables the Efficient Biosynthesis of Extender Units for Engineering Polyketide Carbon Scaffolds. ACS Catal. 2021, 11, 12179–12185. [Google Scholar]
80.
Zheng M, Zhang J, Zhang W, Yang L, Yan X, Tian W, et al. An Atypical Acyl-CoA Synthetase Enables Efficient Biosynthesis of Extender Units for Engineering a Polyketide Carbon Scaffold. Angew. Chem. Int. Ed. 2022, 61, e202208734. [Google Scholar]
81.
Koryakina I, Kasey C, McArthur JB, Lowell AN, Chemler JA, Li S, et al. Inversion of Extender Unit Selectivity in the Erythromycin Polyketide Synthase by Acyltransferase Domain Engineering. ACS Chem. Biol. 2017, 12, 114–123. [Google Scholar]
82.
Li Y, Zhang W, Zhang H, Tian W, Wu L, Wang S, et al. Structural Basis of a Broadly Selective Acyltransferase from the Polyketide Synthase of Splenocin. Angew. Chem. Int. Ed. 2018, 57, 5823–5827. [Google Scholar]
83.
Reid R, Piagentini M, Rodriguez E, Ashley G, Viswanathan N, Carney J, et al. A Model of Structure and Catalysis for Ketoreductase Domains in Modular Polyketide Synthases. Biochemistry 2003, 42, 72–79. [Google Scholar]
84.
Jez JM, Bowman ME, Noel JP. Expanding the Biosynthetic Repertoire of Plant Type III Polyketide Synthases by Altering Starter Molecule Specificity. Proc. Natl. Acad. Sci. USA 2002, 99, 5319–5324. [Google Scholar]
85.
Morita H, Yamashita M, Shi S-P, Wakimoto T, Kondo S, Kato R, et al. Synthesis of Unnatural Alkaloid Scaffolds by Exploiting Plant Polyketide Synthase. Proc. Natl. Acad. Sci. USA 2011, 108, 13504–13509. [Google Scholar]
86.
Zhou Y, Tao W, Qi Z, Wei J, Shi T, Kang Q, et al. Structural and Mechanistic Insights into Chain Release of the Polyene PKS Thioesterase Domain. ACS Catal. 2022, 12, 762–776. [Google Scholar]
87.
Wang Z, Bagde SR, Zavala G, Matsui T, Chen X, Kim C-Y. De Novo Design and Implementation of a Tandem Acyl Carrier Protein Domain in a Type I Modular Polyketide Synthase. ACS Chem. Biol. 2018, 13, 3072–3077. [Google Scholar]
88.
Wang H, Liang J, Yue Q, Li L, Shi Y, Chen G, et al. Engineering the Acyltransferase Domain of Epothilone Polyketide Synthase to Alter the Substrate Specificity. Microb. Cell Factor. 2021, 20, 86. [Google Scholar]
89.
Klaus M, Buyachuihan L, Grininger M. Ketosynthase Domain Constrains the Design of Polyketide Synthases. ACS Chem. Biol. 2020, 15, 2422–2432. [Google Scholar]
90.
Englund E, Schmidt M, Nava AA, Lechner A, Deng K, Jocic R, et al. Expanding Extender Substrate Selection for Unnatural Polyketide Biosynthesis by Acyltransferase Domain Exchange within a Modular Polyketide Synthase. J. Am. Chem. Soc. 2023, 145, 8822–8832. [Google Scholar]
91.
Rowe CJ, Böhm IU, Thomas IP, Wilkinson B, Rudd BAM, Foster G, et al. Engineering a Polyketide with a Longer Chain by Insertion of an Extra Module into the Erythromycin-Producing Polyketide Synthase. Chem. Biol. 2001, 8, 475–485. [Google Scholar]
92.
Nielsen ML, Isbrandt T, Petersen LM, Mortensen UH, Andersen MR, Hoof JB, et al. Linker Flexibility Facilitates Module Exchange in Fungal Hybrid PKS-NRPS Engineering. PLoS ONE 2016, 11, e0161199. [Google Scholar]
93.
Kim M-S, Cho W-J, Song MC, Park S-W, Kim K, Kim E, et al. Engineered Biosynthesis of Milbemycins in the Avermectin High-Producing Strain Streptomyces Avermitilis. Microb. Cell Factor. 2017, 16, 9. [Google Scholar]
94.
Yi D, Agarwal V. Biosynthesis-Guided Discovery and Engineering of α-Pyrone Natural Products from Type I Polyketide Synthases. ACS Chem. Biol. 2023, 18, 1060–1065. [Google Scholar]
95.
Zhao C, Huang Y, Guo C, Yang B, Zhang Y, Lan Z, et al. Heterologous Expression of Spinosyn Biosynthetic Gene Cluster in Streptomyces Species Is Dependent on the Expression of Rhamnose Biosynthesis Genes. J. Mol. Microbiol. Biotechnol. 2017, 27, 190–198. [Google Scholar]
96.
Anderson TM, Clay MC, Cioffi AG, Diaz KA, Hisao GS, Tuttle MD, et al. Amphotericin Forms an Extramembranous and Fungicidal Sterol Sponge. Nat. Chem. Biol. 2014, 10, 400–406. [Google Scholar]
97.
Pandey RP, Sohng JK. Glycosyltransferase-Mediated Exchange of Rare Microbial Sugars with Natural Products. Front. Microbiol. 2016, 7, 1849. [Google Scholar]
98.
Pandey RP, Parajuli P, Sohng JK. Metabolic Engineering of Glycosylated Polyketide Biosynthesis. Emerg. Top. Life Sci. 2018, 2, 389–403. [Google Scholar]
99.
Qiao L, Dong Y, Zhou H, Cui H. Effect of Post–Polyketide Synthase Modification Groups on Property and Activity of Polyene Macrolides. Antibiotics 2023, 12, 119. [Google Scholar]
100.
Trefzer A, Hoffmeister D, Künzel E, Stockert S, Weitnauer G, Westrich L, et al. Function of Glycosyltransferase Genes Involved in Urdamycin A Biosynthesis. Chem. Biol. 2000, 7, 133–142. [Google Scholar]
101.
Hoffmeister D, Wilkinson B, Foster G, Sidebottom PJ, Ichinose K, Bechthold A. Engineered Urdamycin Glycosyltransferases Are Broadened and Altered in Substrate Specificity. Chem. Biol. 2002, 9, 287–295. [Google Scholar]
102.
Hong JSJ, Park SH, Choi CY, Sohng JK, Yoon YJ. New Olivosyl Derivatives of Methymycin/Pikromycin from an Engineered Strain of Streptomyces Venezuelae. FEMS Microbiol. Lett. 2004, 238, 391–399. [Google Scholar]
103.
Zhang X, Li S. Expansion of Chemical Space for Natural Products by Uncommon P450 Reactions. Nat. Prod. Rep. 2017, 34, 1061–1089. [Google Scholar]
104.
Lee SK, Basnet DB, Hong JSJ, Jung WS, Choi CY, Lee HC, et al. Structural Diversification of Macrolactones by Substrate-Flexible Cytochrome P450 Monooxygenases. Adv. Synth. Catal. 2005, 347, 1369–1378. [Google Scholar]
105.
Chen S, Zhang C, Zhang L. Investigation of the Molecular Landscape of Bacterial Aromatic Polyketides by Global Analysis of Type II Polyketide Synthases. Angew. Chem. Int. Ed. 2022, 61, e202202286. [Google Scholar]
106.
Nava AA, Roberts J, Haushalter RW, Wang Z, Keasling JD. Module-Based Polyketide Synthase Engineering for de Novo Polyketide Biosynthesis. ACS Synth. Biol. 2023, 12, 3148–3155. [Google Scholar]
107.
Medema MH, Breitling R, Bovenberg R, Takano E. Exploiting Plug-and-Play Synthetic Biology for Drug Discovery and Production in Microorganisms. Nat. Rev. Microbiol. 2011, 9, 131–137. [Google Scholar]
108.
Zhang X, Wang Y, Zhang Y, Wang M. CRISPR/Cas9-Mediated Multi-Locus Promoter Engineering in Ery Cluster to Improve Erythromycin Production in Saccharopolyspora Erythraea. Microorganisms 2023, 11, 623. [Google Scholar]
109.
Wang X, Ning X, Zhao Q, Kang Q, Bai L. Improved PKS Gene Expression With Strong Endogenous Promoter Resulted in Geldanamycin Yield Increase. Biotechnol. J. 2017, 12, 1700321. [Google Scholar]
110.
Wei P-L, Fan J, Yu J, Ma Z, Guo X, Keller NP, et al. Quantitative Characterization of Filamentous Fungal Promoters on a Single-Cell Resolution to Discover Cryptic Natural Products. Sci. China Life Sci. 2023, 66, 848–860. [Google Scholar]
111.
Tu R, Zhang Y, Hua E, Bai L, Huang H, Yun K, et al. Droplet-Based Microfluidic Platform for High-Throughput Screening of Streptomyces. Commun. Biol. 2021, 4, 647. [Google Scholar]
112.
Tu R, Li S, Li H, Wang M. Advances and applications of droplet-based microfluidics in evolution and screening of engineered microbial strains. Syn. Bio. J. 2023, 4, 165–184. [Google Scholar]
113.
Zhang P, Wang H, Xu H, Wei L, Liu L, Hu Z, et al. Deep Flanking Sequence Engineering for Efficient Promoter Design Using DeepSEED. Nat. Commun. 2023, 14, 6309. [Google Scholar]
114.
Horbal L, Siegl T, Luzhetskyy A. A Set of Synthetic Versatile Genetic Control Elements for the Efficient Expression of Genes in Actinobacteria. Sci. Rep. 2018, 8, 491. [Google Scholar]
115.
Jeong Y, Kim J-N, Kim MW, Bucca G, Cho S, Yoon YJ, et al. The Dynamic Transcriptional and Translational Landscape of the Model Antibiotic Producer Streptomyces Coelicolor A3(2). Nat. Commun. 2016, 7, 11605. [Google Scholar]
116.
Bai C, Zhang Y, Zhao X, Hu Y, Xiang S, Miao J, et al. Exploiting a Precise Design of Universal Synthetic Modular Regulatory Elements to Unlock the Microbial Natural Products in Streptomyces. Proc. Natl. Acad. Sci. USA 2015, 112, 12181–12186. [Google Scholar]
117.
Yi JS, Kim MW, Kim M, Jeong Y, Kim E-J, Cho B-K, et al. A Novel Approach for Gene Expression Optimization through Native Promoter and 5′ UTR Combinations Based on RNA-Seq, Ribo-Seq, and TSS-Seq of Streptomyces Coelicolor. ACS Synth. Biol. 2017, 6, 555–565. [Google Scholar]
118.
Zhao M, Wang S-L, Tao X-Y, Zhao G-L, Ren Y-H, Wang F-Q, et al. Engineering Diverse Eubacteria Promoters for Robust Gene Expression in Streptomyces Lividans. J. Biotechnol. 2019, 289, 93–102. [Google Scholar]
119.
Goshisht MK. Machine Learning and Deep Learning in Synthetic Biology: Key Architectures, Applications, and Challenges. ACS Omega 2024, 9, 9921–9945. [Google Scholar]
120.
Guo W, Xiao Z, Huang T, Zhang K, Pan H-X, Tang G-L, et al. Identification and Characterization of a Strong Constitutive Promoter stnYp for Activating Biosynthetic Genes and Producing Natural Products in Streptomyces. Microb. Cell Factor. 2023, 22, 127. [Google Scholar]
121.
Cho MK, Lee BT, Kim HU, Oh M. Systems Metabolic Engineering of Streptomyces Venezuelae for the Enhanced Production of Pikromycin. Biotechnol. Bioeng. 2022, 119, 2250–2260. [Google Scholar]
122.
Wang X, Zhang C, Wang M, Lu W. Genome-Scale Metabolic Network Reconstruction of Saccharopolyspora Spinosa for Spinosad Production Improvement. Microb. Cell Factor. 2014, 13, 41. [Google Scholar]
123.
Kim M, Yi JS, Lakshmanan M, Lee D-Y, Kim B-G. Transcriptomics-Based Strain Optimization Tool for Designing Secondary Metabolite Overproducing Strains of Streptomyces Coelicolor. Biotechnol. Bioeng. 2016, 113, 651–660. [Google Scholar]
124.
Song C, Luan J, Cui Q, Duan Q, Li Z, Gao Y, et al. Enhanced Heterologous Spinosad Production from a 79-Kb Synthetic Multi-Operon Assembly. ACS Synth. Biol. 2019, 8, 137–147. [Google Scholar]
125.
Jiang C, Zhou H, Sun H, He R, Song C, Cui T, et al. Establishing an Efficient Salinomycin Biosynthetic Pathway in Three Heterologous Streptomyces Hosts by Constructing a 106-Kb Multioperon Artificial Gene Cluster. Biotechnol. Bioeng. 2021, 118, 4668–4677. [Google Scholar]
126.
Shao Z, Rao G, Li C, Abil Z, Luo Y, Zhao H. Refactoring the Silent Spectinabilin Gene Cluster Using a Plug-and-Play Scaffold. ACS Synth. Biol. 2013, 2, 662–669. [Google Scholar]
127.
Hou P, Woolner VH, Bracegirdle J, Hunt P, Keyzers RA, Owen JG. Stictamycin, an Aromatic Polyketide Antibiotic Isolated from a New Zealand Lichen-Sourced Streptomyces Species. J. Nat. Prod. 2023, 86, 526–532. [Google Scholar]
128.
Hochrein L, Machens F, Gremmels J, Schulz K, Messerschmidt K, Mueller-Roeber B. AssemblX: A User-Friendly Toolkit for Rapid and Reliable Multi-Gene Assemblies. Nucleic Acids Res. 2017, 45, e80. [Google Scholar]
129.
Mukherjee M, Caroll E, Wang ZQ. Rapid Assembly of Multi-Gene Constructs Using Modular Golden Gate Cloning. J. Vis. Exp. 2021, 168, e61993. [Google Scholar]
130.
Liu R, Deng Z, Liu T. Streptomyces Species: Ideal Chassis for Natural Product Discovery and Overproduction. Metab. Eng. 2018, 50, 74–84. [Google Scholar]
131.
Nepal KK, Wang G. Streptomycetes: Surrogate Hosts for the Genetic Manipulation of Biosynthetic Gene Clusters and Production of Natural Products. Biotechnol. Adv. 2019, 37, 1–20. [Google Scholar]
132.
Feng J, Hauser M, Cox RJ, Skellam E. Engineering Aspergillus Oryzae for the Heterologous Expression of a Bacterial Modular Polyketide Synthase. J. Fungi. 2021, 7, 1085. [Google Scholar]
133.
Vandova GA, O’Brien RV, Lowry B, Robbins TF, Fischer CR, Davis RW, et al. Heterologous Expression of Diverse Propionyl-CoA Carboxylases Affects Polyketide Production in Escherichia Coli. J. Antibiot. 2017, 70, 859–863. [Google Scholar]
134.
Gonzalez-Garcia RA, Nielsen LK, Marcellin E. Heterologous Production of 6-Deoxyerythronolide B in Escherichia Coli through the Wood Werkman Cycle. Metabolites 2020, 10, 228. [Google Scholar]
135.
Kang S-Y, Heo KT, Hong Y-S. Optimization of Artificial Curcumin Biosynthesis in E. Coli by Randomized 5′-UTR Sequences To Control the Multienzyme Pathway. ACS Synth. Biol. 2018, 7, 2054–2062. [Google Scholar]
136.
Liu Z, Xu J, Liu H, Wang Y. Engineered EryF Hydroxylase Improving Heterologous Polyketide Erythronolide B Production in Escherichia Coli. Microb. Biotechnol. 2022, 15, 1598–1609. [Google Scholar]
137.
Bond C, Tang Y, Li L. Saccharomyces Cerevisiae as a Tool for Mining, Studying and Engineering Fungal Polyketide Synthases. Fungal Genet. Biol. 2016, 89, 52–61. [Google Scholar]
138.
Li Y, Lin P, Lu X, Yan H, Wei H, Liu C, et al. Plasmid Copy Number Engineering Accelerates Fungal Polyketide Discovery upon Unnatural Polyketide Biosynthesis. ACS Synth. Biol. 2023, 12, 2226–2235. [Google Scholar]
139.
Zhao M, Zhao Y, Yao M, Iqbal H, Hu Q, Liu H, et al. Pathway Engineering in Yeast for Synthesizing the Complex Polyketide Bikaverin. Nat. Commun. 2020, 11, 6197. [Google Scholar]
140.
Liu Z, Zhu Z, Tang J, He H, Wan Q, Luo Y, et al. RNA-Seq-Based Transcriptomic Analysis of Saccharopolyspora Spinosa Revealed the Critical Function of PEP Phosphonomutase in the Replenishment Pathway. J. Agric. Food Chem. 2020, 68, 14660–14669. [Google Scholar]
141.
Kittikunapong C, Ye S, Magadán-Corpas P, Pérez-Valero Á, Villar CJ, Lombó F, et al. Reconstruction of a Genome-Scale Metabolic Model of Streptomyces Albus J1074: Improved Engineering Strategies in Natural Product Synthesis. Metabolites 2021, 11, 304. [Google Scholar]
142.
Dang L, Liu J, Wang C, Liu H, Wen J. Enhancement of Rapamycin Production by Metabolic Engineering in Streptomyces Hygroscopicus Based on Genome-Scale Metabolic Model. J. Ind. Microbiol. Biotechnol. 2017, 44, 259–270. [Google Scholar]
143.
Gu B, Kim DG, Kim D-K, Kim M, Kim HU, Oh M-K. Heterologous Overproduction of Oviedomycin by Refactoring Biosynthetic Gene Cluster and Metabolic Engineering of Host Strain Streptomyces Coelicolor. Microb. Cell Factor. 2023, 22, 212. [Google Scholar]
144.
Fu S, An Z, Wu L, Xiang Z, Deng Z, Liu R, et al. Evaluation and Optimization of Analytical Procedure and Sample Preparation for Polar Streptomyces Albus J1074 Metabolome Profiling. Synth. Syst. Biotechnol. 2022, 7, 949–957. [Google Scholar]
145.
Li J, Mu X, Dong W, Chen Y, Kang Q, Zhao G, et al. A Non-Carboxylative Route for the Efficient Synthesis of Central Metabolite Malonyl-CoA and Its Derived Products. Nat. Catal. 2024, 7, 361–374. [Google Scholar]
146.
He H, Tang J, Chen J, Hu J, Zhu Z, Liu Y, et al. Flaviolin-Like Gene Cluster Deletion Optimized the Butenyl-Spinosyn Biosynthesis Route in Saccharopolyspora Pogona. ACS Synth. Biol. 2021, 10, 2740–2752. [Google Scholar]
147.
Rang J, Li Y, Cao L, Shuai L, Liu Y, He H, et al. Deletion of a Hybrid NRPS-T1PKS Biosynthetic Gene Cluster via Latour Gene Knockout System in Saccharopolyspora Pogona and Its Effect on Butenyl-spinosyn Biosynthesis and Growth Development. Microb. Biotechnol. 2021, 14, 2369–2384. [Google Scholar]
148.
Ye S, Magadán-Corpas P, Pérez-Valero Á, Villar CJ, Lombó F. Metabolic Engineering Strategies for Naringenin Production Enhancement in Streptomyces Albidoflavus J1074. Microb. Cell Factor. 2023, 22, 167. [Google Scholar]
149.
Yin S, Wang X, Shi M, Yuan F, Wang H, Jia X, et al. Improvement of Oxytetracycline Production Mediated via Cooperation of Resistance Genes in Streptomyces Rimosus. Sci. China Life Sci. 2017, 60, 992–999. [Google Scholar]
150.
Lei C, Wang J, Liu Y, Liu X, Zhao G, Wang J. A Feedback Regulatory Model for RifQ-Mediated Repression of Rifamycin Export in Amycolatopsis Mediterranei. Microb. Cell Factor. 2018, 17, 14. [Google Scholar]
151.
Shan Y, Guo D, Gu Q, Li Y, Li Y, Chen Y, et al. Genome Mining and Homologous Comparison Strategy for Digging Exporters Contributing Self-Resistance in Natamycin-Producing Streptomyces Strains. Appl. Microbiol. Biotechnol. 2020, 104, 817–831. [Google Scholar]
152.
Vasanthakumar A, Kattusamy K, Prasad R. Regulation of Daunorubicin Biosynthesis in Streptomyces Peucetius—Feed Forward and Feedback Transcriptional Control: Regulation of Daunorubicin Biosynthesis in Streptomyces Peucetius. J. Basic Microbiol. 2013, 53, 636–644. [Google Scholar]
153.
Li W, Sharma M, Kaur P. The DrrAB Efflux System of Streptomyces Peucetius Is a Multidrug Transporter of Broad Substrate Specificity. J. Biol. Chem. 2014, 289, 12633–12646. [Google Scholar]
154.
Malla S, Niraula NP, Liou K, Sohng JK. Self-Resistance Mechanism in Streptomyces Peucetius: Overexpression of drrA, drrB and drrC for Doxorubicin Enhancement. Microbiol. Res. 2010, 165, 259–267. [Google Scholar]
155.
Qiu J, Zhuo Y, Zhu D, Zhou X, Zhang L, Bai L, et al. Overexpression of the ABC Transporter AvtAB Increases Avermectin Production in Streptomyces Avermitilis. Appl. Microbiol. Biotechnol. 2011, 92, 337–345. [Google Scholar]
156.
Chu L, Li S, Dong Z, Zhang Y, Jin P, Ye L, et al. Mining and Engineering Exporters for Titer Improvement of Macrolide Biopesticides in Streptomyces. Microb. Biotechnol. 2022, 15, 1120–1132. [Google Scholar]
157.
Ji C-H, Kim H, Kang H-S. Synthetic Inducible Regulatory Systems Optimized for the Modulation of Secondary Metabolite Production in Streptomyces. ACS Synth. Biol. 2019, 8, 577–586. [Google Scholar]
158.
Palmer CM, Miller KK, Nguyen A, Alper HS. Engineering 4-Coumaroyl-CoA Derived Polyketide Production in Yarrowia Lipolytica through a β-Oxidation Mediated Strategy. Metab. Eng. 2020, 57, 174–181. [Google Scholar]
159.
Tian J, Yang G, Gu Y, Sun X, Lu Y, Jiang W. Developing an Endogenous Quorum-Sensing Based CRISPRi Circuit for Autonomous and Tunable Dynamic Regulation of Multiple Targets in Streptomyces. Nucleic Acids Res. 2020, 48, 8188–8202. [Google Scholar]
160.
Qiu S, Yang A, Zeng H. Flux Balance Analysis-Based Metabolic Modeling of Microbial Secondary Metabolism: Current Status and Outlook. PLoS Comput. Biol. 2023, 19, e1011391. [Google Scholar]
161.
Wang X, Chen N, Cruz-Morales P, Zhong B, Zhang Y, Wang J, et al. Elucidation of Genes Enhancing Natural Product Biosynthesis through Co-Evolution Analysis. Nat. Metab. 2024, 6, 933–946. [Google Scholar]
162.
Hirsch M, Desai RR, Annaswamy S, Keatinge-Clay AT. Mutagenesis Supports AlphaFold Prediction of How Modular Polyketide Synthase Acyl Carrier Proteins Dock With Downstream Ketosynthases. Proteins Struct. Funct. Bioinforma. 2024, 1–10. doi: 10.1002/prot.26733.