
Alterations of T Cell Subsets Associated with Sickle Cell Trait
Author Information
Other Information
1
Department of Epidemiology, University of Washington, Seattle, WA 98105, USA
2
Public Health Sciences Division, Fred Hutchinson Cancer Research Center, Seattle, WA 98105, USA
3
Department of Genetics, University of North Carolina, Chapel Hill, NC 27599, USA
4
Jackson Heart Study, Jackson, MS 39216, USA
5
Department of Pathology and Laboratory Medicine, University of Vermont Larner College of Medicine, Burlington, VT 05405, USA
6
Division of Biostatistics, Institute for Health and Equity, and Cancer Center, Medical College of Wisconsin, Milwaukee, WI 53226, USA
*
Authors to whom correspondence should be addressed.
Received: 22 July 2024 Accepted: 27 September 2024 Published: 08 October 2024

© 2024 The authors. This is an open access article under the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/).
Blood Genomics
2025,
9(1), 10001;
DOI: 10.70322/bgd.2025.10001
ABSTRACT:
Sickle cell trait (SCT) has been associated with alterations
in various immune-related laboratory parameters including lower circulating
lymphocyte counts. To further characterize the impact of SCT on the immune
system, we performed flow cytometry of monocyte and lymphocyte immune cell
subsets from peripheral blood mononuclear cells collected in a large,
community-based cohort of SCT-positive (n = 68) and SCT-negative (n = 959) Black adults. SCT was significantly associated with lower
proportions of CD8+ and CD4+ T cell subsets that
include senescent-like markers of repeated immune system challenges. These
immune alterations could have potential implications for the susceptibility of
individuals with SCT to various infectious diseases.
Keywords:
Sickle Cell; T Lymphocyte; Flow Cytometry; CD4; CD8
1. Introduction
Sickle cell trait (SCT) is characterized by the inheritance of a single copy of the Hb S mutation and affects, among other populations, approximately 10% of Black individuals in the United States [1,2]. While SCT traditionally has been considered a benign carrier state, it has become increasingly apparent that individuals with SCT may experience subclinical low-grade hemolysis and tend to have lower red cell-related laboratory values (e.g., hemoglobin, red cell count, mean corpuscular volume (MCV)) compared to age-and race-matched Hb A/A individuals [3]. In addition, SCT has been associated with an increased risk of various conditions including exertional rhabdomyolysis [4], chronic kidney disease (CKD) [5], diabetes [6], and venous thromboembolic disease [7]. In particular, SCT has been associated with several kidney complications, including impaired urinary concentration, hematuria/papillary necrosis, lower eGFR, albuminuria, and higher risk of chronic kidney disease (CKD) and progression to end-stage kidney disease (ESKD) [8]. More recently, SCT has been associated with alterations in various immune-related parameters including higher absolute counts and proportion of neutrophils, lower lymphocyte counts and proportion [9], and higher circulating levels of inflammatory proteins such as fractalkine/CX3CL1 [10]. These immune alterations could have potential implications for the recently reported associations of SCT with susceptibility to various infectious diseases [11,12,13,14,15]. To further characterize the impact of SCT on the adaptive and innate immune responses, we performed flow cytometry of monocyte and lymphocyte immune cell subsets from peripheral blood mononuclear cells (PBMC) collected in a large, community-based cohort of SCT-positive and SCT-negative Black adults.
2. Methods
The Jackson Heart Study (JHS) is a community-based, longitudinal cohort study designed to identify CVD risk factors among self-identified Black or African American adults age ≥ 21 years from the Jackson, Mississippi metropolitan area, as previously described [16]. Flow cytometry was performed using peripheral blood mononuclear cells (PBMCs) isolated from blood collected and cryopreserved from 1028 JHS participants during the baseline examination (2000–2004). All study sites obtained informed consent and institutional review board approval.
2.1. Data Collection
Baseline socio-demographic and medical history were collected using standardized questionnaires. To determine sickle cell mutation status, genotyping for HBB rs334 was derived from TOPMed whole genome sequencing (WGS), as previously described [17]. Of 1028 JHS participants with flow cytometry data, there were 68 with SCT (genotyped as Hb A/S) and 959 genotyped as Hb A/A. One individual was genotyped as Hb S/S and was excluded from further analysis. African genetic ancestry was estimated from WGS as the top genotype principal component (PC) using PC-AiR [18]. A complete blood cell count (CBC) and leukocyte differential were performed during the baseline exam using an automated electronic cell counter. Serum creatinine was measured at baseline using an enzymatic method that was traceable to an isotope dilution mass spectrometry reference creatinine standard. Glomerular filtration rate was estimated (eGFR) from serum creatinine using the CKD Epidemiology Collaboration (CKD-EPI) equation [19].
2.2. Flow Cytometry
Cell phenotyping assay methods, reagents, and flow cytometry gating strategies for lymphocytes and monocytes have been described in detail [18]. In general, cryopreserved cells were thawed, washed, stained for viability and surface labeled for most phenotypes. For regulatory cells, the surface labeled cells were further explored by intracellular staining for transcription factors. For stimulation assays (i.e., Th1, Th2, Th17), surface-labeled cells were stimulated with PMA/Ionomycin to determine intracellular production of cytokines. Data were collected on a Miltenyi MACSQuant 16 flow cytometer using single-color controls to set compensation and isotype controls to set negative gates and analyzed using FCS Express software. Immune cell phenotypes were expressed as proportions of their parent populations. Details on all the phenotypes measured, their parent populations and their gating strategies were as previously described [20].
2.3. Statistical Analysis
To assess the association of immune cell (lymphocyte or monocyte) subset proportions with SCT (Hb A/S) versus non-SCT African American controls (Hb A/A), we performed linear regression with rank-based normal transformed cell proportion as the dependent variable and SCT as the independent variable, adjusting for baseline age, sex, and African genetic ancestry. In total, 141 different immune cell phenotypes were evaluated for association with SCT (Supplemental Table S1). Notably, many of the T cell, B cell, and monocyte phenotypes exhibit varying degrees of correlation with one another. Therefore, to adjust for multiple testing and control the family-wise error rate at an alpha = 0.05, we utilized a resampling-based method that implicitly accounts for the dependence structure of the individual test statistics [21]. Within the JHS flow cytometry dataset, there were 164 families with two or more members, encompassing 793 of the 1027 participants. Therefore, we also conducted regression analyses accounting for familial relationships using mixed linear models assuming a correlation structure within families. Since there were no substantive differences between the results of regression models accounting for family structure versus those not accounting for family structure, we present the results of the latter. In additional sensitivity analyses, we added other covariates (hemoglobin, eGFR, total lymphocyte count) to the SCT-immune cell phenotype association regression models to assess whether the associations are robust to additional adjustment for phenotypes known to be associated with SCT status.
3. Results
Among the 1027 participants included in the study, 48% were female and the average age was 62 years (range 40 to 88 years). There were 68 with SCT and 959 Black participants without SCT. The baseline characteristics of the participants are shown in Table 1, stratified by SCT status. Compared to Black individuals without SCT (Hb A/A), those with SCT (HbA/S) had lower age- and sex-adjusted estimated glomerular filtration rate (eGFR), MCV, total white blood cell (WBC) count, and total lymphocyte count (all p < 0.05).
In age-, sex-, and genetic ancestry-adjusted linear regression models, SCT was significantly associated with lower proportions of CD8+ and CD4+ T cell subsets that included CD57+, CD27−, and/or CD28− (all senescent-like markers of repeated immune system challenges) and the CD8+Granzyme B+ cells (all adjusted p < 0.05) (Table 2).
There was little evidence of association between SCT and proportions of B cells, T regulatory cells, CD4+ or CD8+ stimulated cells, gamma delta T cells, natural killer cells, or monocyte subsets (Supplemental Table S2). The full set of results and their comparison when accounting for versus not accounting for family structure is shown in Supplemental Table S2. In sensitivity analyses, none of the significant T cell subset-SCT associations were altered by additional adjustment for other phenotypes known to be associated with SCT status (hemoglobin, eGFR, and total lymphocyte count) (Table 2). A heat map of pairwise correlation coefficients between the 12 significant SCT-associated T cell phenotypes shows two main clusters consisting of CD4+ versus CD8+ subsets (Figure 1).
Spearman pairwise correlations are shown for each of the twelve CD4+ or CD8+ lymphocyte subsets significantly associated with sickle cell trait in Table 2.
Table 1. Baseline characteristics of study participants.
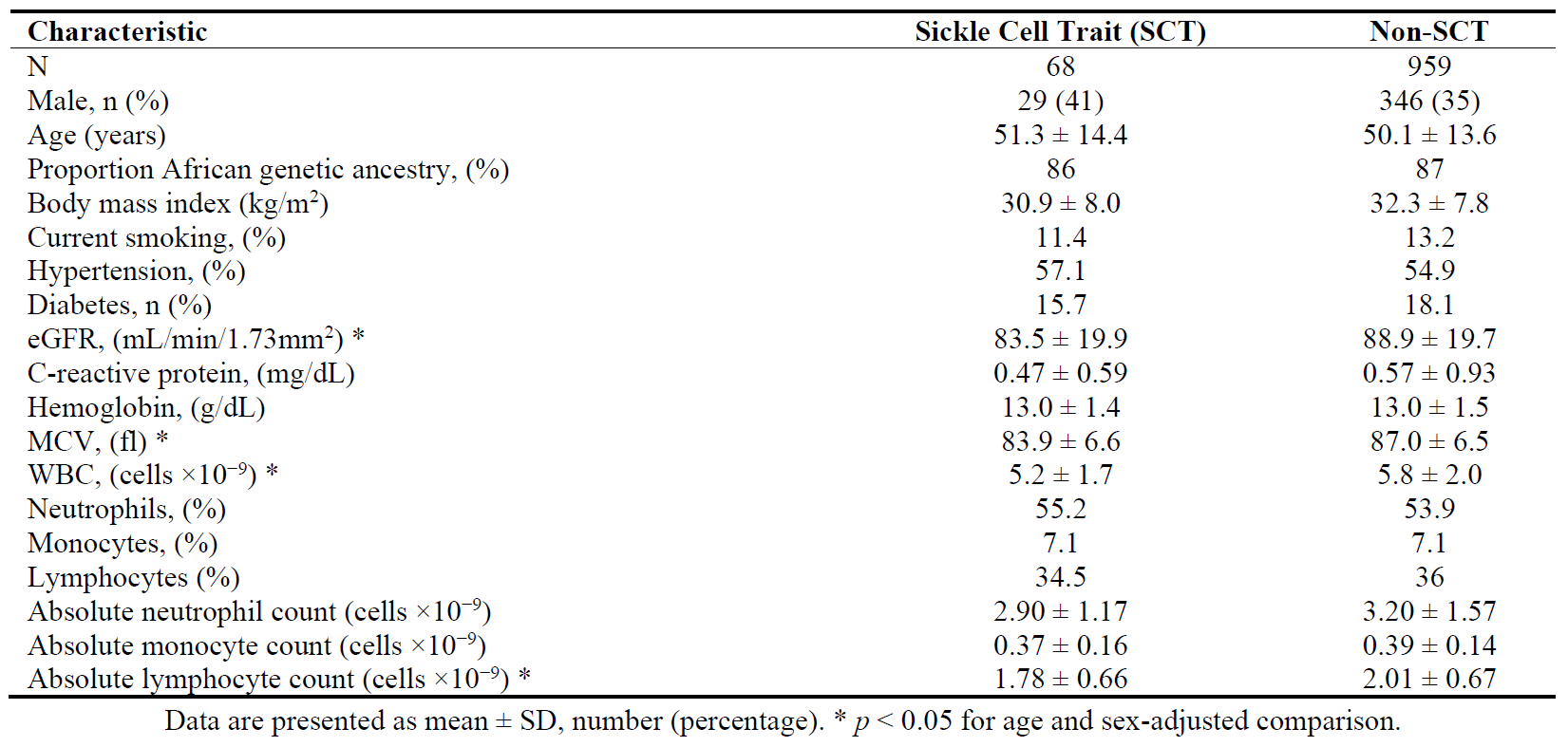
Table 2. Significant associations of sickle cell trait with immune cell phenotypes.

Figure 1. Heatmap of pairwise correlations between SCT-associated lymphocyte subsets.
4. Discussion
Our current analysis was motivated by the recent finding that SCT is associated with lower total circulating lymphocyte count [9]. The reduced proportions of senescent and differentiated or proliferative CD4+ and CD8+ T cells with effector functions we observed among individuals with SCT could be due to reduced proliferative capacity, activation, and/or reduced survival. The overall pattern we observed in this community-based African American sample appears to suggest a state of immune inactivation. These immune alterations may have implications for the clinical consequences and pathobiology of SCT, as discussed further below.
Among individuals with SCT, chronic low-grade hemolysis may lead to saturation of heme scavenging mechanisms, resulting in elevated serum levels of free heme. Extracellular heme can trigger a type 1 interferon inflammatory response and activate the NLRP3 inflammasome, which can have downstream consequences for both innate and adaptive immune systems [22,23]; these include recruitment of innate immune cells to sites of inflammation, polarization of macrophages to the pro-inflammatory state, production of interferon-γ, and modulation of T cell responses. Through modulation of heme oxygenase-1 expression, chronic hemolysis may additionally inhibit effector T-cell responses [22].
The pathophysiology and molecular mechanisms underlying the association of SCT with various clinical sequelae remain incompletely understood. Local conditions within the renal medulla and vasa recta (low oxygen tension, reduced blood flow, acidosis, and hyperosmolarity) can promote HbS polymerization and sickling, which likely contribute to SCT susceptibility to kidney disorders. Dysregulation of the adaptive immune system is a common feature of kidney diseases in general, including progression to ESKD [24]. Therefore, the circulating T cell alterations observed in current study may in part reflect local tissue damage in the kidneys and additionally play a role in SCT-related nephropathy. Moreover, the SCT-associated adaptive immune alterations (e.g., reduced proportions of T effector cells) may contribute to the susceptibility of SCT individuals to infectious diseases. SCT has been associated with increased risk of COVID-19 [11], post-operative infection [14], and pneumonia [15]. Future studies that assess the impact of modifiers of Hb S polymerization (such as Hb F levels) [25] on immune function and susceptibility to infection among individuals with SCT may be warranted.
The immune system also has been recognized to play an important role in pathophysiology of sickle cell disease (SCD) and its complications [26]. The release of free heme during intravascular hemolysis leads to release of proinflammatory cytokines, activation of innate immunity, and chronic inflammation, all of which contribute to vaso-occlusive crises [27]. Compared to the innate immune system, changes in adaptive immune response in SCD have been less well characterized. Alterations in T cell subsets have been reported in relatively small numbers of SCD individuals, either in the steady-state or during vaso-occlusive crises, with conflicting results [28,29,30,31]. Interestingly, a pattern of alterations in T cell subsets including reduced CD28− effector T cells similar to that observed in the current SCT study was reported in a group of SCD patients in acute crisis state compared to steady-state SCD patients [29]. In contrast, another study reported higher CD4+CD28− differentiated effector memory cells among steady-state SCD patients compared to controls and an association of higher CD4+CD28− cells in SCD patients with a history of vaso-occlusive crises [31]. Additional investigation of the adaptive immune response and the relationship of lymphocyte phenotypes to clinical outcomes among individuals with either SCD or SCT may identify therapeutic targets for vaso-occlusive crises and susceptibility to infection.
Supplementary Materials
The following supporting information can be found at: https://www.sciepublish.com/article/pii/297, Table S1: Immune phenotype characterization; Table S2: Association of sickle cell trait with immune cell phenotypes, adjusting for age, sex, and genetic ancestry caption.
Acknowledgments
The authors wish to thank the staff and participants of the Jackson Heart Study.
Author Contributions
Ethics Statement
The study was conducted according to the guidelines of the Declaration of Helsinki and approved by the Institutional Review Board of the University of Washington (STUDY00000450) on 26 January 2024.
Informed Consent Statement
Funding
This research was supported by funding from Jackson Heart Study (JHS) is supported and conducted in collaboration with Jackson State University (HHSN268201800013I), Tougaloo College (HHSN268201800014I), the Mississippi State Department of Health (HHSN268201800015I/HHSN26800001) and the University of Mississippi Medical Center (HHSN268201800010I, HHSN268201800011I, and HHSN268201800012I) contracts from the National Heart, Lung, and Blood Institute and the National Institute for Minority Health and Health Disparities (NIMHD). Genome sequencing for “NHLBI TOPMed: The Jackson Heart Study” (phs000964.v1.p1) was performed at the Northwest Genomics Center (HHSN268201100037C). Core support, including centralized genomic read mapping and genotype calling, along with variant quality metrics and filtering were provided by the TOPMed Informatics Research Center (3R01HL-117626-02S1; contract HHSN268201800002I). Core support including phenotype harmonization, data management, sample-identity QC, and general program coordination were provided by the TOPMed Data Coordinating Center (R01HL-120393; U01HL-120393; contract HHSN268201800001I). LMR is supported by R01AG075884. The flow cytometric analysis in JHS participants was supported by R01HL132947. The views expressed in this manuscript are those of the authors and do not necessarily represent the views of the National Heart, Lung, and Blood Institute; the National Institutes of Health; or the U.S. Department of Health and Human Services.
Declaration of Competing Interest
L.M.R. is a consultant for the TOPMed Administrative Coordinating Center (through Westat).
References
1.
Xu JZ, Thein SL. The carrier state for sickle cell disease is not completely harmless. Haematologica 2019, 104, 1106–1111. [Google Scholar]
2.
Naik RP, Smith-Whitley K, Hassell KL, Umeh NI, de Montalembertet M, Sahota P, et al. Clinical Outcomes Associated With Sickle Cell Trait: A Systematic Review. Ann. Intern. Med. 2018, 169, 619–627. [Google Scholar]
3.
Raffield LM, Ulirsch JC, Naik RP, Lessard S, Handsaker RE, Jain D, et al. Common α-globin variants modify hematologic and other clinical phenotypes in sickle cell trait and disease. PLoS Genet. 2018, 14, e1007293. [Google Scholar]
4.
Nelson DA, Deuster PA, Carter R 3rd, Hill OT, Wolcott VL, Kurina LM. Sickle Cell Trait, Rhabdomyolysis, and Mortality among U.S. Army Soldiers. N. Engl. J. Med. 2016, 375, 435–442. [Google Scholar]
5.
Naik RP, Derebail VK, Grams ME, Franceschini N, Auer PL, Peloso GM, et al. Association of sickle cell trait with chronic kidney disease and albuminuria in African Americans. JAMA 2014, 312, 2115–2125. [Google Scholar]
6.
Hulsizer J, Resurreccion WK, Shi Z, Wei J, Ladson-Gary S, Zheng SL, et al. Sickle cell trait and risk for common diseases: Evidence from the UK Biobank. Am. J. Med. 2022, 135, e279–e287. [Google Scholar]
7.
Folsom AR, Tang W, Roetker NS, Kshirsagar AV, Derebail VK, Lutsey PL, et al. Prospective study of sickle cell trait and venous thromboembolism incidence. J. Thromb Haemost. 2015, 13, 2–9. [Google Scholar]
8.
Ataga KI, Saraf SL, Derebail VK. The nephropathy of sickle cell trait and sickle cell disease. Nat. Rev. Nephrol. 2022, 18, 361–377. [Google Scholar]
9.
Mikhaylova AV, McHugh CP, Polfus LM, Raffield LM, Boorgula PM, Blackwell TW, et al. Whole-genome sequencing in diverse subjects identifies genetic correlates of leukocyte traits: The NHLBI TOPMed program. Am. J. Hum. Genet. 2021, 108, 1836–1851. [Google Scholar]
10.
Cai Y, Franceschini N, Surapaneni A, Garrett ME, Tahir UA, Hsu L, et al. Differences in the circulating proteome in individuals with versus without sickle cell trait. Clin. J. Am. Soc. Nephrol. 2023, 18, 1416–1425. [Google Scholar]
11.
Michelon I, Vilbert M, Pinheiro IS, Costa IL, Lorea CF, Castonguay M, et al. COVID-19 outcomes in patients with sickle cell disease and sickle cell trait compared with individuals without sickle cell disease or trait: a systematic review and meta-analysis. EClinicalMedicine 2023, 66, 102330. [Google Scholar]
12.
Nekhai S, Kumari N. HIV-1 infection in sickle cell disease and sickle cell trait: role of iron and innate response. Expert Rev. Hematol. 2022, 15, 253–263. [Google Scholar]
13.
Kumari N, Nouraie M, Ahmad A, Lassiter H, Khan J, Diaz S, et al. Restriction of HIV-1 infection in sickle cell trait. Blood Adv. 2021, 5, 4922–4934. [Google Scholar]
14.
Waters TL, Wilder JH, Ross BJ, Salas Z, Sherman WF. Effect of sickle cell trait on total hip arthroplasty in a matched cohort. J. Arthroplasty. 2022, 37, 892–896.e5. [Google Scholar]
15.
Chen HH, Shaw DM, Petty LE, Graff M, Bohlender RJ, Polikowsky HG, et al. Host genetic effects in pneumonia. Am. J. Hum. Genet. 2021, 108, 194–201. [Google Scholar]
16.
Taylor HA Jr, Wilson JG, Jones DW, Sarpong DF, Srinivasan F, Garrison RJ, et al. Toward resolution of cardiovascular health disparities in African Americans: design and methods of the Jackson Heart Study. Ethn Dis. 2005, 15 (4 Suppl 6), S6-4–17. [Google Scholar]
17.
Hu Y, Stilp AM, McHugh CP, Rao S, Jain D, Zheng X, et al. Whole-genome sequencing association analysis of quantitative red blood cell phenotypes: The NHLBI TOPMed program. Am. J. Hum. Genet. 2021, 108, 874–893. [Google Scholar]
18.
Conomos MP, Miller MB, Thornton TA. Robust inference of population structure for ancestry prediction and correction of stratification in the presence of relatedness. Genet. Epidemiol. 2015, 39, 276–293. [Google Scholar]
19.
Levey AS, Stevens LA. Estimating GFR using the CKD Epidemiology Collaboration (CKD-EPI) creatinine equation: more accurate GFR estimates, lower CKD prevalence estimates, and better risk predictions. Am. J. Kidney Dis. 2010, 55, 622–627. [Google Scholar]
20.
Fang Y, Doyle MF, Chen J, Mez J, Satizabal CL, Alosco ML, et al. Circulating immune cell phenotypes are associated with age, sex, CMV, and smoking status in the Framingham Heart Study offspring participants. Aging (Albany NY) 2023, 15, 3939–3966. [Google Scholar]
21.
Romano JP, Wolf M. Control of generalized error rates in multiple testing. Ann. Stat. 2007, 35, 1378–1408. [Google Scholar]
22.
Zhong H, Yazdanbakhsh K. Hemolysis and immune regulation. Curr. Opin. Hematol. 2018, 25, 177–182. [Google Scholar]
23.
Salgar S, Bolívar BE, Flanagan JM, Anum SJ, Bouchier-Hayes L. The NLRP3 inflammasome fires up heme-induced inflammation in hemolytic conditions. Transl. Res. 2023, 252, 34–44. [Google Scholar]
24.
Tecklenborg J, Clayton D, Siebert S, Coley SM. The role of the immune system in kidney disease. Clin. Exp. Immunol. 2018, 192, 142–150. [Google Scholar]
25.
Steinberg MH, Embury SH. Alpha-thalassemia in blacks: genetic and clinical aspects and interactions with the sickle hemoglobin gene. Blood 1986, 68, 985–990. [Google Scholar]
26.
de Azevedo JTC, Malmegrim KCR. Immune mechanisms involved in sickle cell disease pathogenesis: current knowledge and perspectives. Immunol Lett. 2020, 224, 1–11. [Google Scholar]
27.
Kato GJ, Steinberg MH, Gladwin MT. Intravascular hemolysis and the pathophysiology of sickle cell disease. J. Clin. Invest. 2017, 127, 750–760. [Google Scholar]
28.
Marchesani S, Bertaina V, Marini O, Cossutta M, Di Mauro M, Rotulo GA, et al. Inflammatory status in pediatric sickle cell disease: Unravelling the role of immune cell subsets. Front. Mol. Biosci. 2022, 9, 1075686. [Google Scholar]
29.
Patel S, Chandrakar D, Wasnik PN, Nayak S, Shah S, Nanda R, et al. Altered T-cell profile in sickle cell disease. Biomark Med. 2023, 17, 241–252. [Google Scholar]
30.
Balandya E, Reynolds T, Obaro S, Makani J. Alteration of lymphocyte phenotype and function in sickle cell anemia: Implications for vaccine responses. Am. J. Hematol. 2016, 91, 938–946. [Google Scholar]
31.
ElAlfy MS, Adly AAM, Ebeid FSE, Eissa DS, Ismail EAR, Mohammed YH, et al. Immunological role of CD4+CD28null T lymphocytes, natural killer cells, and interferon-gamma in pediatric patients with sickle cell disease: relation to disease severity and response to therapy. Immunol Res. 2018, 66, 480–490. [Google Scholar]