As mentioned above, the four Notch receptors share a similar basic structure but differ in several aspects. The Notch receptors have a similar extracellular domain, which contains three lin-12/Notch /LNR) motifs, 29–36 EGF-like repeats, and a heterodimerization domain. While Notch1 and Notch 2 have 36 EGF-like repeats, Notch 3 has 34 and Notch4 29, resulting in Notch3 having a shorter ECD than Notch1 and Notch2 but longer than Notch4 ECD [
14] (). Moreover, Notch3 and Notch4 lack the transactivation domain (TAD) found in the N1ICD and N2ICD (A). In contrast, Notch3 has a unique PPXY motif, known to act as a WW domain recognition site for the endocytic regulator WWP2 (B). Furthermore, Notch4 lacks the Notch cytokine response (NCR) region present in the other Notch receptors [
15] (). All of these structural differences in the Notch receptors may account for the functional differences observed among them. Interestingly, structural data has shown that Notch3 seems to be the most easily cleaved of all Notch receptors [
16]. In fact, data shows that it can be cleaved and activated even in the absence of a ligand [
17]. Furthermore, Notch3 has specific mutational hotspots that are associated with the development of Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy (CADASIL), a hereditary neurological disorder characterized by vascular abnormalities in the brain [
18]. This is the major implication of Notch3 dysfunction in human disease. In addition, it has been shown to be involved in several cancers including breast, lung, pancreatic, and colorectal cancer [
19], and in fibrotic disease in distinct tissues, which is described in detail next.
3.1. Notch3 in Liver Fibrosis
Continuous liver injury may lead to extensive liver fibrosis, also known as cirrhosis, which is a major cause of death worldwide [
20]. The Notch pathway plays an important role in liver development and regeneration [
21]; therefore, a dysregulation in this signaling may give rise to the development of pathology.
Hepatic stellate cells (HSCs) are a type of resident mesenchymal cell that has features of both resident fibroblasts and pericytes and produces an extracellular matrix (ECM) [
22]. HSCs activation into proliferative, fibrogenic myofibroblasts is considered the central driver of hepatic fibrosis in experimental and human liver injury [
23]. In fact, single-cell RNA sequencing (scRNAseq) studies have shown that HSCs are the main source of myofibroblasts during cirrhosis [
24]. Macrophages are found in close proximity to activated HSCs and have been demonstrated to play a key role in fibrosis initiation and progression [
25,
26]. Both Kupffer cells (resident macrophages) and recruited macrophages produce pro-fibrotic mediators such as transforming growth factor β (TGFβ) and platelet-derived growth factor (PDGF), cytokines and chemokines that further lead to worsened fibrosis [
26,
27]. Interestingly, Notch signaling has been previously shown to be crucial for HSC activation and macrophage polarization, determining the fate of liver fibrogenesis [
28].
The specific implication of Notch3 signaling in liver fibrosis has been previously described by Chen et al. [
29]. They found that the Notch3 pathway could regulate the activation of HSCs. They observed by immunohistochemistry that all fibrotic liver tissues from patients with chronic active hepatitis were positive for Notch3, whereas Notch3 was not detected in normal liver tissues. Futhermore, they showed that overexpression of Notch3 led to increased expression of α-SMA and collagen I in HSC-T6 cells, while downregulation of Notch3 yielded the opposite effects. Consistently, the expression of
Notch3 has been found to be upregulated in diseased human livers and a rat model of liver fibrosis [
30,
31]. In contrast, in a study using a mouse model of liver fibrosis, the authors could not detect changes in Notch3, but rather an increase in the expression and activation of Notch1 and Notch2 [
32]. However, multi-lineage modelling of ligand and receptor interactions has revealed Notch3 signaling as an important regulator of mesenchymal cell function within the human liver fibrotic niche [
33]. This study, describes the interaction between Notch ligands derived from scar-associated endothelial cells and Notch3+ scar-associated mesenchymal cells. Indeed, coculture of primary human HSCs and endothelial cells from cirrhotic liver promoted fibrillar collagen production by HSCs, which was inhibited using a Notch-signaling inhibitor. Accordingly, the knockdown of Notch3 expression in primary human HSCs resulted in reduced fibrillar collagen expression. In addition, Cong and Liu et al. reported that Notch3 expression at the mRNA and protein levels is significantly higher in patients with moderate and severe liver fibrosis compared with patients without liver fibrosis. In contrast, no significant difference existed between mild patients and patients without liver fibrosis [
34]. This study, also showed that the fibrogenic factors such as α-SMA, collagen I, TGF-β1 and Smad3 were significantly increased upon Notch3 upregulation in a human HSC line. Accordingly, the opposite effect was observed when Notch3 was downregulated. In addition, the Notch target gene
Hes1 was increased in liver biopsies from patients with nonalcoholic steatohepatitis (NASH)/fibrosis [
32], suggesting a potential implication of the Notch3/Hes1 axis in hepatic fibrosis.
Based on the ample evidence of the regulatory role of the Notch pathway in liver fibrosis, its modulation emerges as a potential novel therapy for cirrhotic patients to improve symptoms or even revert the disease.
3.2. Notch3 in Kidney Fibrosis
Chronic kidney disease (CKD) affects more than 10% of the world’s population, and its progression may lead to renal fibrosis [
35]. Notch3 is considered one of the major receptors involved in renal fibrosis [
36] despite the fact that few studies have established its precise role in this context. Djudjaj et al. revealed in 2012 the likely critical implication of Notch3 in kidney fibrosis since genetic deletion of Notch3 using global Notch3-knockout mice protected them from tubulointerstitial fibrosis induced by unilateral ureteral obstruction (UUO), a murine model of kidney fibrosis, exhibiting significantly lower collagen deposition and reduced number of α-SMA-positive cells [
37]. Furthermore, the Notch target genes
HeyL,
Hes5, and
Hey2 were shown to be potentially downstream of Notch3 signaling in this context. In a study by Xiao et al. [
38], the authors showed that the expression of Notch 1, 3, and 4, Notch intracellular domain (NICD), and its target genes Hes1 and HeyL were upregulated in mice with UUO. Importantly, pharmacological inhibition using the pan-Notch inhibitor DBZ resulted in a reduction of pathological fibroblasts and other fibrotic parameters, indicating the relevant implication of Notch signaling in kidney fibrogenesis.
In addition to myofibroblasts, which are considered the major cellular player in the development and progression of kidney fibrosis, other cell types also contribute; this is the case of tubule epithelial cells, in which sustained activation of the Notch pathway has a key role in fibrogenesis [
39]. Interstitial inflammation and fibrosis are one of the major pathological changes of polycystic kidney disease (PKD), a genetic disorder affecting millions of people worldwide [
40]. A recent study observed that Notch3 was highly upregulated in patients with PKD [
41]. These authors showed that chronic epithelial overexpression of N3ICD in mice resulted in severe tubulointerstitial inflammation and fibrosis that progressed with time. Interestingly, Notch3 deficiency may also predispose to kidney fibrosis, as indicated by anecdotal reports from CADASIL patients [
42] and research involving angiotensin II-infused mice [
43]. By contrast, one study shows that the global deletion of Notch3 did not protect mice from fibrosis, claiming that
Notch3 minimally contributes to CKD [
44]. A recent study showed that Notch3 expression on CD45+ leucocytes regulates immune cell infiltration following UUO [
45]. Interestingly, using chimeric animal models, fibrosis still ensued despite the lack of prominent leukocyte infiltrates. However, using global Notch3 KO mice showed a reduction in renal fibrosis, demonstrating that Notch3 activation in kidney cells is crucial for the development of kidney fibrosis regardless of the presence of leukocyte infiltrates.
All these observations support a model in which the Notch3 pathway is reactivated in response to kidney injury and contributes to the fibrotic response, suggesting that decreasing Notch3 activity in CKD patients could potentially be used to ameliorate disease development.
3.3. Notch3 in Skin Fibrosis
Skin fibrosis has a wide variety of pathological manifestations such as hypertrophic scarring, keloid formation, chronic cutaneous Graft-versus-Host-Disease (GvDH), nephrogenic fibrosing dermopathy or systemic sclerosis (SSc) [
46]. Nevertheless, the exact mechanisms leading to fibrotic skin conditions remain to be elucidated.
In vivo experiments using a skin incision model have shown that RBPJk KO mice exhibit a reduced expression of several collagens and fibrotic markers in the healed skin [
47]. Consistently, pharmacological Notch inhibition by treatment with the gamma secretase inhibitor DAPT reduced bleomycin-induced fibrosis in a dose-dependent manner with significant decreases in dermal thickening, numbers of myofibroblasts and hydroxyproline content [
48]. However, to our knowledge, there are no existing studies investigating specifically the direct effect of Notch3 signaling on the development or progression of skin fibrosis. A study focused on hair outgrowth and skin fibrosis revealed that the expression of
Notch3 was elevated in αSMA+ dermal sheath cells when βcatenin was overexpressed, alongside the expression of fibrotic markers such as
Ctgf,
Col1a1, and
Col1a2 [
49].
Specific
NOTCH3 polymorphisms correlate with the susceptibility to diffuse cutaneous systemic sclerosis (DcSSc) [
50]. Treatment of human neutrophils, dermal microvascular endothelial cells, and primary skin fibroblasts with dSsc neutrophil-derived exosomes induced the expression of Notch pathway genes and fibrotic-related genes [
51]. Recessive dystrophic epidermolysis bullosa (RDEB) is a rare skin fragility disorder caused by mutations in COL7A1, and progressive fibrosis is one of its major hallmarks. RDEB-derived fibroblasts treated with DAPT showed a reduction of fibrotic traits. However, in this case, it is probably due to the blockade of the Notch1 receptor rather than Notch3 due to the observed upregulation of the first one in these cells [
52].
Interestingly, Notch3 has been demonstrated to induce terminal differentiation of keratynocites [
53], the major cell type of the epidermis, and this could be used in treatment strategies focused on tissue repair. Overall, the general lack of studies of Notch signaling in skin fibrosis shows that there is plenty of scope for the study of Notch3 signaling in this pathology.
3.4. Notch3 in Heart Fibrosis
Cardiovascular disease is the leading cause of death worldwide, and cardiac fibrosis is a common pathophysiological manifestation of most cardiovascular diseases [
54,
55]. To our knowledge, the first observation of the potential role of Notch3 in cardiac fibrosis was reported in 2012 [
56]. Here, they observed that apelin-13, a specific ligand for the angiotensin-like 1 receptor, upregulated the expression of several genes, including
Notch3, and attenuated cardiac fibrosis in infarcted mice. This was accompanied by increased bone marrow cells (BMCs) recruitment. In addition, the treatment of cultured BMCs with apelin in vitro increased
Notch3 expression [
57]. Further, treatment with bone marrow cells overexpressing apelin in vivo, significantly increased angiogenesis and attenuated cardiac fibrosis formation in post-MI mice, although whether this effect is mediated by Notch3 remains unknown [
57].
A recent study reported that
Notch3−/− mice exhibit left ventricular hypertrophy combined with mild fibrosis [
58]. Furthermore, it was shown that
Notch3 was expressed in vascular smooth muscle cells and pericytes and not in cardiomyocites, thereby suggesting that the observed phenotype could be due to a perturbation of Notch3 signaling in these cells. In line with this, Chen et al. indicated that a reduction in Notch3 may disrupt endothelial cell-pericyte communication, causing pericyte detachment that promotes its differentiation into myofibroblasts during ischemia-reperfusion or myocardial infarction [
59]. However, the exact mechanism by which Notch3 may be implicated in the development of cardiac fibrosis was not assessed [
60].
Interestingly, a previous study showed that Notch3 overactivation in cardiac fibroblasts in vitro promotes apoptosis and inhibits proliferation, fibroblast to myofibroblast differentiation, and ECM production, while Notch3 downregulation shows the opposite effects [
60]. Accordingly, overactivating Notch3 signaling in vivo further prevented miocardial infarction-induced cardiac fibrosis in rats [
60]. Consistently, a similar study performed in mice demonstrated that Notch3 signaling inhibits TGF-β1/Smad3 signaling in cardiac fibroblasts, impairing fibroblast-to-myofibroblast transition, evidenced by a decrease in αSMA and Type I collagen expression [
61]. Altogether, in contrast to the pathogenic role of Notch3 signaling upregulation observed in fibrosis in other organs, evidence support Notch3 downregulation as a driver of fibrosis in the heart and its activation as a therapeutic option to treat cardiac fibrosis.
3.5. Notch3 in Pancreatic Fibrosis
Pancreatic fibrosis is a characteristic feature of chronic pancreatitis and pancreatic cancer [
62]. Even though, up to date, there is no direct association between Notch3 signaling and pancreatic fibrosis nor any published work focused on this topic, a couple of studies have observed a relation between
Notch3 and chronic pancreatitis [
63,
64]. First, transcripts of
Notch3 in microdissected ectatic ducts of chronic pancreatitis were found to be elevated [
63]. In a recent study, the authors identified
Notch3 as a risk gene and promising biomarker for chronic pancreatitis, which needs further confirmation in a clinical study [
64]. Pancreatic cancer is one of the most lethal cancer types, and its incidence is increasing [
65]. Pancreatic ductal adenocarcinoma (PDAC) is the most common type of pancreatic cancer and is characterized by the presence of abundant desmoplastic stroma. Desmoplasia is the formation of dense fibrotic tissue within and around tumor tissue, which is generated by activated fibroblasts, myofibroblasts, pancreatic stellate cells and tumor cells. It comprises 80% to 90% of the tumor volume [
66]. Activated pancreatic stellate cells (PaSCs) are the key cellular source of cancer-associated fibroblasts in the pancreatic stroma of PDAC patients. Interestingly, Song et al. showed that Notch3 is overexpressed in both human stromal cells from PDAC patients and activated mouse PaSCs [
67]. Remarkably, upon
Notch3 knockdown in vitro, a decrease in both RNA and protein levels of the fibrotic markers αSMA, collagen I, and fibronectin, and also of the Notch target gene Hes1, was observed. Furthermore, cell proliferation and migration were reduced when Notch3 was downregulated [
68]. All these findings suggest that Notch3 regulates cell migration, proliferation and expression of fibrotic markers in the stromal component of pancreatic cancer. Importantly,
Notch3 expression inversely correlates with response to therapy and overall survival in patients with pancreatic cancer treated with gemcitabine [
68], highlighting the relevant implication of this pathway in the disease. Thus, targeting Notch3 could impact pancreatic tumorigenesis by limiting fibrosis and emerge as a novel therapy for PDAC patients.
3.6. Notch3 in Lung Fibrosis
Idiopathic pulmonary fibrosis (IPF) is the most prevalent lung fibrotic disease within the interstitial lung diseases, a group of more than 200 parenchymal pulmonary disorders mainly characterized by alveolar damage and interstitial inflammation and/or fibrosis. IPF is a chronic and progressive pulmonary disease, and the fact that there is currently no cure, a lack of treatments impeding its progression and that patients are not always eligible to receive a lung transplant often leads to death in 3–5 years post-diagnosis. One of the histopathological features of IPF is the presence of fibroblasts foci, which are active sites of remodelling in the fibrotic lung where pathological fibroblasts produce and secrete excessive amounts of ECM proteins and are lined by aberrant epithelial cells not normally found in healthy lungs and with an important role in tissue fibrosis.
A study from 2010 showed that myofibroblast differentiation was impaired in vivo in Notch2
−/−; Notch3
−/− double mutant embryos but not in single mutants in the developing lung mesenchyme, suggesting the redundant function of these two receptors inducing myofibroblast differentiation [
69]. In the adult lung, Notch1 plays an essential role in fibroblast-to-myofibroblast differentiation in the pathogenesis of pulmonary fibrosis, as reported by Hu et al. [
70]. Interestingly, a previous publication identifies a unique Pdgfrβ+ population defined by Notch3 as a discriminating marker to emerge in the fibrotic mouse lung [
71]. Furthermore, an upregulation of Notch3 in fibroblasts was observed by scRNA sequencing comparing healthy and fibrotic human lungs [
72]. Our previous work demonstrates the specific role of Notch3 in fibroblast-to-myofibroblast differentiation [
73]. First, we observed that around 60% of the collagen-expressing cells showed Notch3 activity in the mouse lung at homeostasis. After bleomycin-induced lung injury, almost half of the emerging αSMA+ myofibroblasts showed Notch3 activity, suggesting a role of Notch3 in fibrogenesis. To test this hypothesis, we used a
Notch3 KO mouse line and observed that Notch3 deficiency attenuated bleomycin-induced lung fibrosis and impeded lung function decline. Furthermore, we demonstrated that Notch3 signaling regulates fibroblast survival and myofibroblast differentiation. Consistently, a previous study showed that ROS-dependent activation of p38, JNK1/2, and Notch3 activation in IMR-90 cells (primary human lung fibroblasts) promoted basal and TGFβ1-induced differentiation and expression of ECM proteins in vitro [
74]. Moreover, treatment with the Notch inhibitor DAPT or Notch3-specific siRNA suppressed the expression of αSMA. Using immunohistochemistry, the authors found Notch3 in the cytoplasm, membrane, and nuclei of myofibroblasts and in epithelial cells of IPF lungs, while they did not detect it in healthy lungs. In contrast, we detected Notch3 activity in αSMA+ myofibroblasts of the healthy lung and observed an expansion of the N3ICD+ myofibroblasts in the IPF lung [
73]. All in all, targeting Notch3 signaling could be a potential strategy to tackle IPF progression.
Filtering for “Clinical Trial” or “Randomized Controlled Trial” and searching for articles containing both terms “Notch3” and “fibrosis,” yields zero results using PubMed as of September 2024. Similarly, when entering “fibrosis” as a condition/disease and “NOTCH inhibitor” as intervention/treatment in “clinicaltrials.gov”, no results are obtained. However, when searching just for “NOTCH inhibitor”, regardless of the condition/disease, a total of 50 studies are listed, suggesting that there is a significant unexplored venue for the establishment of new or repurposed drugs targeting Notch signaling for the treatment of fibrotic diseases.
The use of Notch inhibitors to treat cancer is extensive. Currently, two recruiting clinical trials use pan-NOTCH inhibitors in cancer (NCT05774899, NCT04973683). There are also three additional active studies with the same scope that are not recruiting (NCT02069730, NCT03785964, NCT04871282), which are also focused on the use of pan-NOTCH inhibitors in cancer and, interestingly, the two last are the only clinical trials in phase III. The rest of the clinical trials are also solely focused on treating different cancer diseases and are in phase I or II, with no clinical trials in phase IV. Of note, 22 of the 50 total trials have been terminated, meaning the study was stopped earlier and will not start again. While some of the studies did not specify the reason for termination (NCT00100152, NCT01088763, NCT01071564, NCT01193881, NCT01192763, NCT01238133, NCT01208441), others did: “Business reason” (NCT03422679), “Non-safety reason, business objectives have changed” (NCT01986218), “Administratively Complete” (NCT01151449, NCT01120275, NCT01269411), “Drug was no longer available” (NCT01200810), “Sponsor’s decision” (NCT04461600), “Study drug production halted” (NCT01193868), “stopped prematurely by Company due to decision to terminate all CTEP supplied drug for further development of RO4929097” (NCT01122901), “Company decided to stop the development of drug” (NCT01189240), “The trial was closed because the sponsor became insolvent” (NCT03740100), and “Slow accrual coupled with discontinuation of study drug” (NCT01217411). Furthermore, the use of general Notch inhibitors often leads to adverse effects, which poses a huge problem in the establishment of these drugs as approved disease treatments.
There are some drugs related to Notch ligands that are currently being tested. For instance, two T-cell engagers targeting DLL3, a canonical Notch ligand, are being tested by Boehringer Ingelheim (BI764532) and Merck (HPN328) to treat lung cancer. Unfortunately, to our knowledge, no drugs that specifically inhibit Notch3 signaling are being tested.
The use of Notch3 neutraling antibodies has been proposed in several studies to treat different diseases. In vitro, α-Notch3 (R&D Systems, Minneapolis, MN, USA) has also been used to study the role of Notch3 in collagen II-specific T helper type 1 (Th1) and Th17-type expansion, proposing that selective inhibition of Notch signaling transduced by Notch3 could be used for the treatment of rheumatoid arthritis. More than a decade ago, the development of anti-Notch3 monoclonal antibodies to either block (A4 and A8) or activate (A45 and A79) Notch3 pathway was already a reality [
75]. In a study from Machuca-Parra and Bigger-Allen et al., the authors use an anti-human Notch3 agonist antibody (A13, Genentech) that could be useful for patients with hypomorphic CADASIL mutations in Notch3 and also for others with small vessel disease conditions mechanistically linked to Notch3 loss of function [
76]. Lateral meningocele syndrome (LMS) is a genetic disorder associated with
NOTCH3 mutations, and Yu et al. successfully reversed the skeletal phenotype of LMS in male mice using an anti-Notch3 antibody (Genentech) [
77]. A recent study used an anti-Notch3 antibody (Ab 28042, AVEO Pharmaceuticals), able to bind to both human and murine Notch3, as a drug treatment to reverse pulmonary hypertension (PH) in mouse hypoxia and rat Sugen-hypoxia models of PH [
78]. Interestingly, a novel anti-Notch3 antibody-drug conjugate able to deliver an auristatin-based cytotoxic payload (PF-06650808) reached a phase I study for patients with breast cancer and other advanced solid tumors (NCT02129205). Unfortunately, the study was prematurely terminated as a business decision prior to the start of the dose expansion stage.
Despite promising preclinical results, targeting Notch3 signaling has not reached the clinic yet, but with the advent of new evidence involving Notch3 and disease, the development and use of cell-specific Notch3 inhibitors could be a useful strategy to tackle fibrosis.
Fibrosis is characterized by excessive ECM deposition, which leads to scarring and organ failure. Fibrosis can affect many organs, and 45% of all deaths worldwide are estimated to result from a fibrotic disease. Unfortunately, there are no treatments capable of curing any of them despite the big efforts to understand their physiopathology. Numerous cellular and molecular mechanisms mediate fibrogenesis, such as the Notch pathway, a highly conserved cell-cell communication signaling pathway between neighbouring cells in which physical contact is needed. The role of Notch3 in some diseases is clear, like in CADASIL, where Notch3 mutations are strongly associated with its development. Moreover, the Notch3 receptor has other unique features among the Notch receptors (as shown in 3. Implication of Notch3 in the development of organ fibrosis), and its involvement in fibrogenesis is doubtless.
The use of global Notch3-knockout (KO) mice has allowed us to demonstrate the involvement of Notch3 signaling in the development of fibrosis in the lung and kidney in vivo. However, this tool has a significant caveat: Notch3 signaling is deleted systemically in every cell type and at every stage of development. Since the Notch pathway regulates many different biological processes depending on the cell type and the context, it is difficult to pinpoint the precise role of Notch3 in fibrogenesis using the Notch3 KO mouse line. To solve this problem, conditional Notch3 knockout mice under specific drivers are prime for targeting concrete cell types. In our laboratory, we have already generated various transgenic mouse lines in which we are able to delete Notch3 in a cell-specific manner, giving us the opportunity to accurately explore the role of Notch3 signaling in fibroblast activation.
It would also be interesting to assess Notch3 levels across diseases and cell types on the lookout for new therapeutic windows targeting Notch3 signaling in each fibrotic disease. A relation between upregulated Notch3 mRNA and protein levels with moderate and severe fibrosis stages has been shown in the liver. The fact that no significant relation is observed between mild patients and patients without liver fibrosis shows how the Notch3 pathway could act differently in different stages of fibrotic diseases. However, this is the only example regarding Notch3 levels and fibrotic disease stage and further investigation is needed to establish a clear association.
Despite previous evidence indicating a role for Notch3 in fibrotic disease, targeting Notch3 signaling is an arduous task due to the inherent complexity of the Notch pathway: (1) There are numerous Notch target genes, including the
Hes and
Hey family, and there is no clear pattern to which gene is activated by which NICD, (2) Each receptor may be activated by any Notch ligand and, depending on the ligand, this activation could lead to different molecular outcomes, (3) The Notch pathway can interact with other molecular pathways [
79], (4) Instead of activating them (trans/cis-activation), ligands can also inhibit receptor activation (trans/cis-inhibition) [
80]. All of this partially accounts for the cell and context dependency of Notch signaling and the paradoxical role of Notch3 in fibrotic disease, where both upregulation (liver, kidney, skin, pancreas, and lung) and downregulation (heart) of the pathway can trigger the development of fibrosis.
In general, studying the potential interactions between the Notch3 pathway and other signaling pathways and delving deeply into the mechanism involved in Notch3 activation in each context could shed light on new molecular mechanisms involved in fibrosis development and progression. This could help identify specific target genes associated with the development or progression of a fibrotic disease, which could be valuable in the design of new antifibrotic drugs.
Depending on the cellular and molecular context, Notch3 could regulate proliferation, migration and/or myofibroblast differentiation and play a role in inflammation and tissue regeneration. Therefore, since each cell type could have different roles in the context of injury, targeting Notch3 signaling in a cell-specific fashion is also highly important in the clinical setting. Of note, our previous publication, showed that the decrease in lung fibrosis in Notch3 deficient lungs is not due to a reduction in inflammation, supporting its role in myofibroblast activation [
75]. This also matches the observations made in the kidney, where Notch3 in the inflammatory component was not crucial for the development of fibrosis [
46].
As described above, after binding to a ligand, the NICD is cleaved by a γ-secretase. Many of the clinical trials are focused specifically on the use of γ-secretase inhibitors. Therefore, they are not only inhibiting the signaling of all Notch receptors but also other pathways in which γ-secretases are involved. This and the fact that these treatments are not done in a cell-specific manner could explain why pan-Notch inhibitors often fail, leading to adverse effects.
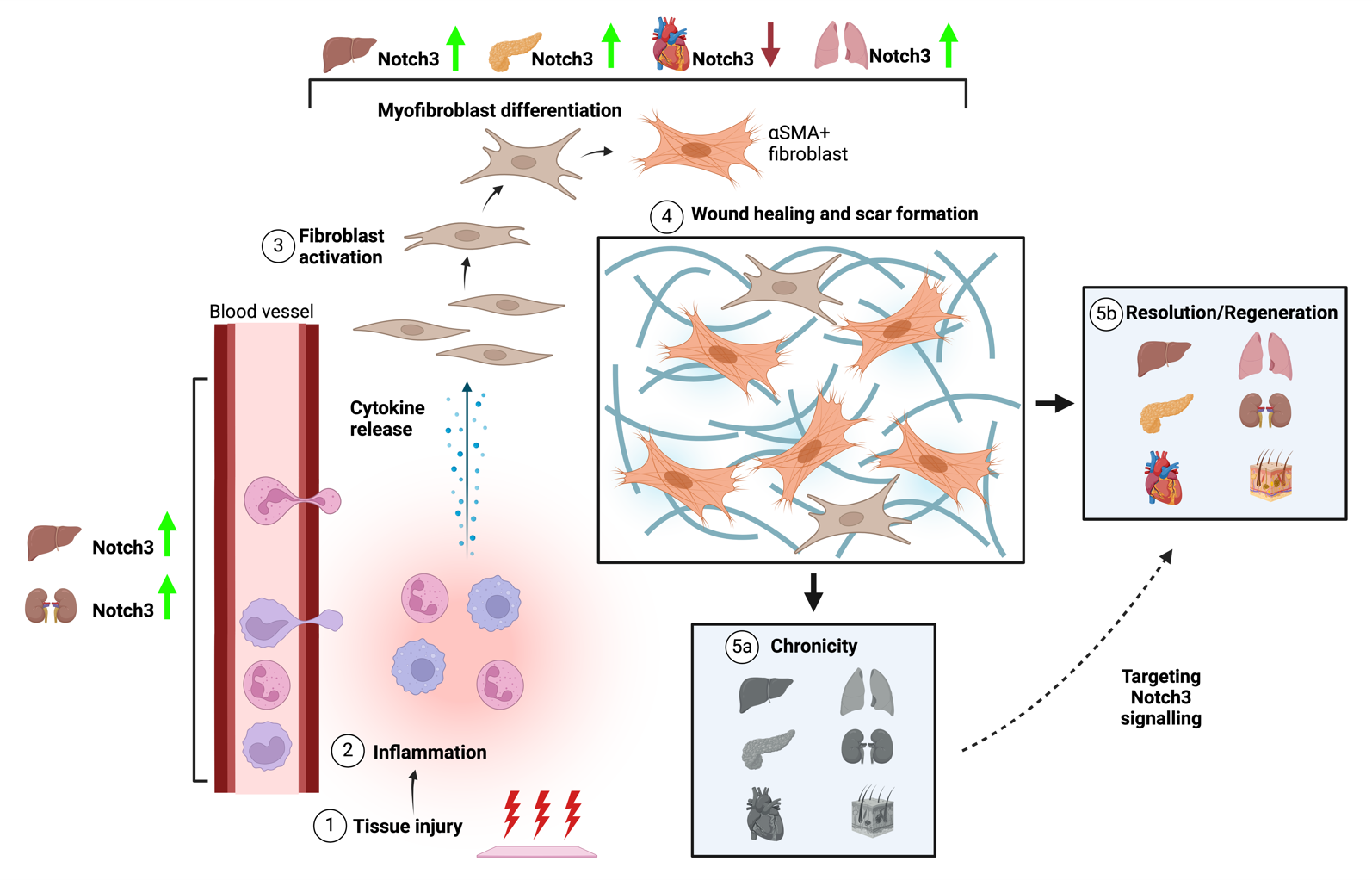
In general, Notch3 plays an important role in tissue fibrogenesis at different levels and in different organs (). Upon tissue injury, there is an immune response followed by the activation of fibroblasts, leading to myofibroblast differentiation and ECM production with the aim of healing the wound and regenerating the tissue. However, dysregulation in this process could lead to chronic fibrosis. The Notch3 could play a pivotal role not only in the process of inflammation but also in myofibroblast differentiation and ECM production in different organs. However, no clear results have been shown in every tissue.
In conclusion, there is still much to do to fully elucidate the implication of Notch3 signaling as a general mechanism in developing fibrosis in different organs. The use of specific mouse transgenic lines targeting Notch3 in specific cell types is extremely important in this matter. Moreover, using anti-Notch3 tools in ex vivo platforms such as precission-cut tissue slices or in vitro using human fibroblasts and organoids will provide decisive results about its potential therapeutic value in human fibrosis. Indeed, we are still far from having an approved anti-Notch3 drug to treat fibrosis, let alone encased in a molecular structure that gives it the capacity to act on the desired cell type in a specific organ. However, this could be the best scenario for the treatment of fibrotic diseases. Therefore, more focus should be placed on the collaboration among disciplines, such as those studying different biological aspects of a disease and those developing new strategies and technologies that could be used to deliver specific drugs targeting specific cells in the desired organ.
. Notch3 contribution to inflammation and fibrosis. The cartoon reflects the different stages from tissue injury to its resolution or its chronicicity. First, an insult to the tissue leads to immune cell recruitment to the site of injury and cytokine release (inflammation). This elicits fibroblast activation and myofibroblast differentiation, which leads to ECM production to give the tissue the scaffold for proper regeneration. However, a dysregulation in any of these steps leads to an excessive scar formation, resulting in the development of chronic fibrotic disease. Despite the scarcity of studies on skin fibrosis, Notch3 upregulation could impact inflammation in the liver and kidneys. Moreover, Notch3 upregulation also promotes tissue fibrosis in the liver, pancreas, and lung, while it seems to have the opposite effect in the heart. All in all, targeting Notch3 signaling could potentially reverse tissue fibrosis and thus give a new opportunity for organ regeneration.
We acknowledge the support of every member of our laboratory for support and feedback on the manuscript.
Conceptualization, A.E.-Z., Z.B.-I. and A.P.-S.; writing—original draft preparation, A.E.-Z.; writing—review and editing, A.E.-Z., Z.B.-I., B.S. and A.P.-S.; supervision, A.P.-S. and B.S. All authors have read and agreed to the published version of the manuscript.
Not applicable.
Not applicable.
This work received no external funding. It was supported by the laboratory of A.P.-S. that is funded by the Institute for Lung Health, Justus Liebig University Giessen (Germany).
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.