
Serine Integrase-based Recombination Enables Direct Plasmid Assembly In Vivo

Author Information
Other Information
School of Physical Science and Technology, ShanghaiTech University, Shanghai 201210, China
*
Authors to whom correspondence should be addressed.
Received: 20 November 2023 Accepted: 19 December 2023 Published: 20 December 2023

© 2023 The authors. This is an open access article under the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/).
Synth. Biol. Eng.
2023,
1(3), 10017;
DOI: 10.35534/sbe.2023.10017
ABSTRACT:
Serine integrases are emerging as one of the powerful tools for synthetic biology. They have been widely developed across genome engineering, biological part construction, genetic circuit design, and in vitro DNA assembly. However, the strategy of in vivo DNA assembly by serine integrases has not yet been reported. To address this opportunity, here we developed a serine integrase-based in vivo DNA (plasmid) assembly approach. First, we demonstrated that the engineered “Acceptor” plasmids could be assembled with diverse “Donor” plasmids by serine integrases (Bxb1 and phiC31) in Escherichia coli (E. coli). Then, by programming the “Donor” plasmids and the host E. coli cells, we established an assembly cascade procedure and finally constructed plasmids that could constitutively express three different fluorescent proteins. Moreover, we used this approach to assemble different chromoprotein genes and generated colored E. coli cells. We anticipate that this approach will enrich the serine integrase-based biotechnology toolbox, and accelerate multiple plasmid assembly for synthetic biology with broad applications.
Keywords:
Serine integrase; Recombinase; Site-specific recombination; Plasmid assembly; Synthetic biology; Biotechnology
1. Introduction
In living cells, DNA plays a core role in regulating the transcription-translation process and programming cellular behaviors. Thus, manipulating DNA can endow cells with novel properties to confer expected characteristics [1]. Over the past two decades, the rapid development of synthetic biology required powerful biotechnologies for DNA manipulation [2,3]. To meet this demand, researchers have focused on the study of nucleases that can recombine/modify DNA. Serine integrases, a class of DNA nucleases that catalyze DNA–DNA site-specific recombination events, have been intensively investigated due to their unique properties of predictability, precision, and high efficiency [4]. Recently, a series of serine integrase-based biotechnologies have been developed [5,6,7], including genome engineering [8,9,10], biological part construction [11,12], genetic circuit design [13,14,15,16], and in vitro DNA assembly [11,17,18,19]. As for in vitro DNA assembly, both linear [17,18,19] and circular [11] DNA could be assembled via serine integrase-based recombination. For example, orthogonal serine integrases could be purified to catalyze the assembly among multiple linear DNA fragments and then generated a whole DNA molecule in vitro. The assembled DNA molecules could be then used for defined applications such as the biosynthesis of carotenoids [19]. Besides, the approach of in vitro circular DNA assembly was performed within two plasmids and purified integrases, and then the assembled plasmids could be transformed into new Escherichia coli (E. coli) strains for co-existing of different genetic circuits [11]. Currently, the approaches of serine integrase-based in vitro DNA assembly have been developed and utilized for broad applications [5]. However, the method of serine integrase-based in vivo DNA (e.g., plasmid) assembly has not yet been reported.
To fill the gap, here we aimed to establish a system in E. coli, which could express serine integrases to facilitate the DNA assembly between two plasmids via site-specific recombination (Figure 1). We first showed that genetically engineered plasmids could be assembled in vivo by two orthogonal serine integrases (Bxb1 and phiC31) [11]. Next, we established a plasmid assembly cascade workflow that could construct more complex plasmids. Furthermore, we utilized this approach to assemble different chromoprotein genes and generated novel colored E. coli cells. Looking forward, we expect that such unique application of serine integrase will be a promising expansion to the genetic manipulation toolbox of synthetic biology.
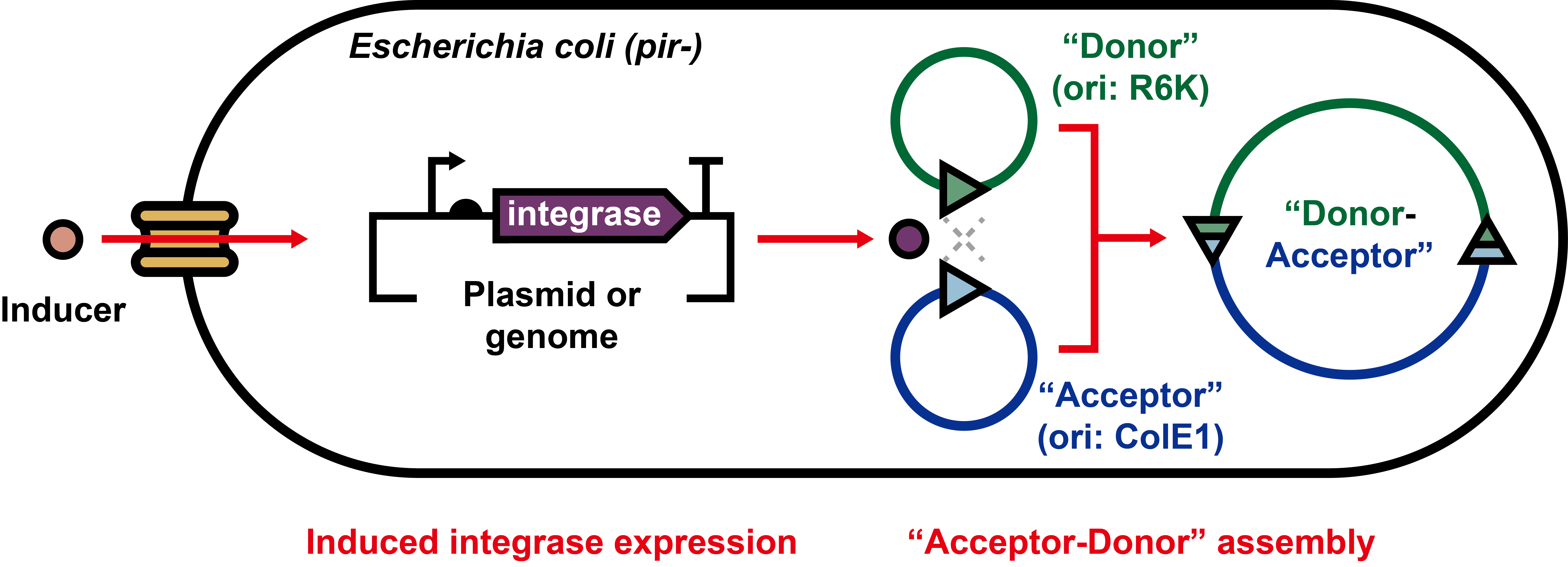
Figure 1. Schematic diagram of serine integrase-based in vivo plasmid assembly system in E. coli. Appropriate inducers are used to induce the expression of integrases. After plasmid assembly, the cells harboring the correctly recombined plasmids are selected by multiple antibiotics-based agar plates. See Figure 2 and Figure S1 for more details.
2. Materials and Methods
2.1. Strains, Genetic Parts, Vectors, and Plasmids
The details of all E. coli strains, genetic parts, vectors, and plasmids used in this study were listed in Tables S1–S4, Supplementary Information. Some genetic parts were derived from our previous works [11,20]. All plasmids were transformed by electroporation with MicroPulser Electroporator (Bio-Rad, Hercules, USA). Competent cells were prepared as described previously [21]. For the electrotransformation, a 50 μL aliquot of E. coli competent cells were mixed with 100 ng plasmid in Gene Pulser/Micropulser Electroporation Cuvettes (0.1 cm gap, Bio-Rad), followed by applying a pulsed voltage value of 1800 V.
2.2. In Vivo Integrase Expression and Western-Blot Analysis
The E. coli DH5α strains containing plasmid (pLW1 for Bxb1, or pLW2 for phiC31) were cultivated in 5 mL LB liquid medium (with 1% (w/v) arabinose) at 30 °C and 250 rpm for 16 h. Note that each culture was inoculated with one fresh colony and the inducer was added at the beginning of the cultivation. After cultivation, cell pellets were collected in 1.5 mL centrifuge tubes at 5000× g and 4 °C for 10 min. The supernatant was discarded and the pellets were resuspended with 1 mL 1× phosphate-buffered saline (pH 7.4) and lysed by sonication (Q125 sonicator, Qsonica, Newtown, USA; 10 s on/off, 50% of amplitude, input energy ~600 Joules). The lysate was then centrifuged at 12,000× g and 4 °C for 10 min. The total (cell lysate) and soluble (supernatant) fractions were separated by SDS-PAGE (Omni-Easy One-Step PAGE Gel Fast Preparation Kit, EpiZyme, Shanghai, China), followed by wet transferring to PVDF membrane (Bio-Rad) with 1× transfer buffer (25 mM Tris-HCl, 192 mM glycine and 20% (v/v) methanol in 1 L ddH2O, pH 8.3). Then, the PVDF membrane was blocked (Protein Free Rapid Blocking Buffer, EpiZyme) for 1 h at room temperature. After washing thrice with TBST for each 5 min, 1:10,000 (TBST-based) diluted HisTag Mouse Monoclonal Antibody (Proteintech, Rosemont, USA) solution was added to the membrane and incubated for 1 h at room temperature. After washing thrice with TBST for each 5 min, 1:10,000 (TBST-based) diluted HRP-Goat Anti-Mouse IgG (H+L) Antibody (Proteintech) solution was added to the membrane and incubated for another 1 h at room temperature. After the last washing with TBST thrice for each 5 min, the membrane was visualized using Omni ECL reagent (EpiZyme) under UVP ChemStudio (analytikjena, Jena, Germany).
2.3. In Vivo Plasmid Assembly Assay
2.3.1. Integrases Were Expressed from “Helper” Plasmids (the First and Second Round of Assembly)
First, the E. coli DH5α strains containing “Acceptor” plasmid (e.g., pLW3) were electroporated with “Helper” plasmid (e.g., pLW1 for Bxb1 expression). Second, the strain was cultivated in 5 mL LB liquid medium (with 1% (w/v) arabinose, ampicillin, and chloramphenicol) at 30 °C and 250 rpm for 16 h. Next day, the strain was electroporated with “Donor1” plasmid (e.g., pLW5) for in vivo “Acceptor-Donor1” assembly, and then separated onto LB-agar plates (with ampicillin, chloramphenicol, and streptomycin) for antibiotic selection (30 °C, at least 16 h). On the third day, the colonies (here the colony number was counted and calculated as assembly efficiency) on LB-agar plates were picked up and inoculated into 5 mL LB liquid medium (with chloramphenicol and streptomycin) for cultivation at 37 °C and 250 rpm for 16 h. On the fourth day, the cell pellets from liquid medium were collected for plasmid extraction, and the extracted plasmids (a plasmid mixture, containing “Helper”, “Acceptor”, “Donor1”, and “Acceptor-Donor1”) were electroporated into wild type E. coli DH5α strain and separated onto LB-agar plates (with chloramphenicol and streptomycin) for cultivation (37 °C, at least 16 h). On the fifth day, the colonies on LB-agar plates were picked up and considered as E. coli strain that solely harboring “Acceptor-Donor1” assembled plasmid.
2.3.2. Integrases Were Expressed from Modified E. coli MG1655 Genome (the Third Round of Assembly)
To release the occupied “AmpR-ampicillin” selection pair in “Helper” plasmid (pLW1 and pLW2), we constructed E. coli MG1655 strains that could express integrases (Bxb1 or phiC31) from the genome rather than “Helper” plasmid. Then, we utilized the strains to assemble (the third round of assembly) the plasmids between “Acceptor-Donor1-Donor2” and “Donor3”.
First, the modified E. coli MG1655 strain (e.g., pLW23 was integrated into the genome for Bxb1 expression) was electroporated with “Acceptor-Donor1-Donor2” plasmid. Second, the strain was cultivated in 5 mL LB liquid medium (with 1% (w/v) arabinose and chloramphenicol) at 37 °C and 250 rpm for 16 h. On the second day, the strain was electroporated with “Donor3” plasmid (e.g., pLW13) for in vivo assembly between “Acceptor-Donor1-Donor2” and “Donor3”, and then separated onto LB-agar plates (with chloramphenicol and ampicillin) for antibiotic selection (37 °C, at least 16 h). On the third day, the colonies on LB-agar plates were picked up and inoculated into 5 mL LB liquid medium (with chloramphenicol and ampicillin) for cultivation at 37 °C and 250 rpm for 16 h. On the fourth day, the cell pellets from liquid medium were collected for plasmid extraction, and the extracted plasmids (a plasmid mixture, containing “Acceptor-Donor1-Donor2”, “Donor3”, and “Acceptor-Donor1-Donor2-Donor3”) were electroporated into wild type E. coli DH5α strain and separated onto LB-agar plates (with chloramphenicol and ampicillin) for cultivation (37 °C, at least 16 h). On the fifth day, the colonies on LB-agar plates were picked up and considered as E. coli strain that solely harboring “Acceptor-Donor1-Donor2-Donor3” assembled plasmid.
2.4. Confocal Fluorescence Microscope Imaging
The E. coli pellets (with fluorescent protein) were collected and resuspended into phosphate-buffered saline (PBS, pH 7.4). Then, the sample was loaded onto microscope slide for imaging. Laser scanning confocal microscopy (FV3000, Olympus, Tokyo, Japan) was used for imaging fluorescence of mCherry (excitation/emission wavelength: 594/610 nm), sfGFP (excitation/emission wavelength: 488/510 nm), and ECFP (excitation/emission wavelength: 405/476 nm), respectively.
2.5. Cultivation and Imaging of E. coli Cells with Chromoproteins
The detailed procedure was described in our previous work [11]. Briefly, 3 mL of each overnight cultivated (37 °C, 250 rpm for 16 h) E. coli DH5α cells was collected in 1.5 mL centrifuge tubes and then the pellets were visualized under bright field. Then, the photos were taken in a mini photo studio using a mobile phone without any image parameter (e.g., brightness, contrast, etc.) adjustments.
3. Results and Discussion
3.1. Serine Integrase-Mediated DNA Assembly Between Two Plasmids
We first sought to express serine integrases in E. coli. To this end, we constructed two “Helper” plasmids that carry two orthogonal integrase genes (Bxb1 and phiC31), which enable E. coli to express these two integrases, respectively, under the control of arabinose induction (Figure 2a). After expression, we performed Western-blot analysis and observed the successful expression of soluble integrases in vivo (Figure 2b). Next, two types of plasmids for assembly were designed as “Acceptor” and “Donor”: (1) The “Acceptor” plasmid consisted of ColE1 ori (origin of replication), antibiotic resistance gene (e.g., CmR–chloramphenicol), and attB (attachment site in bacteria). (2) The “Donor” plasmid contained R6K ori (cannot replicate in E. coli strains that lack gene pir [22], e.g., MG1655 and DH5α), antibiotic resistance gene (e.g., SmR–streptomycin), and attP (attachment site in phage). When the “Acceptor” and “Donor” co-existed in E. coli, the integrase could recombine them to generate an assembled “Acceptor–Donor” plasmid (Figure 2c and Figure S1a). Note that the “Helper” and “Acceptor” plasmids pre-existed in E. coli, serving as a recipient cell. Then, the “Donor” plasmid was electroporated into the recipient for the subsequent recombination. In this process, the assembly might be performed as three different situations. First, the “Donor” plasmid was not successfully electroporated into recipient. Second, the recombination between “Acceptor” and “Donor” was not successful, and the “Donor” plasmid could not normally replicate in recipient (pir-). Third, the recombination between “Acceptor” and “Donor” was successful. Among these three situations, only in the third case, the E. coli strain could normally grow on LB-agar plates (with the selection of two antibiotics) (Figure 2d and Figure S1b). After assembly, the E. coli strain harboring “Acceptor-Donor” plasmid could be picked up for further usages. To demonstrate the approach, we carried out the above-mentioned experiments and the results showed that the assembly between “Acceptor” and “Donor” plasmids was successful (Figure 2e and Figure S1c). Then, we tried to explore the relationship between the recombination efficiency and the arabinose concentration. We found that 1% (w/v) arabinose could facilitate the recombination with 100 ng transformed “Donor” plasmid in the cell, achieving more than 104 CFU (Figure 2f,g). Furthermore, we evaluated the success rate of recombination after a double-antibiotic agar plate selection. The data indicated that both Bxb1- and phiC31-mediated plasmid assembly could reach a 100% success rate (Figure S2). This suggested that the approach of in vivo plasmid assembly between “Acceptor” and “Donor” via serine integrase was feasible and successful.3.2. Establishment of Plasmid Assembly Cascade
After demonstration of DNA assembly between two plasmids, we next inserted three different fluorescent proteins into “Donor” plasmids to endow E. coli with easily observed phenotypes. As expected, the fluorescent E. coli cells could be clearly imaged via microscopy (Figure 3a). Next, to construct more complex plasmids, we attempted to design a plasmid assembly cascade. In the first round of assembly, the “Donor1” was modified and consisted of four parts: R6K ori, antibiotic resistance gene (e.g., SmR), attP (e.g., Bxb1), and attB (e.g., phiC31). When the first round of assembly was finished, the product of “Acceptor–Donor1” plasmid would contain the attB-phiC31 sequence (from “Donor1”). In the second round, the “Donor2” was modified and consisted of four parts: R6K ori, antibiotic resistance gene (e.g., KanR), attP (e.g., phiC31), and attB (e.g., Bxb1). After assembly, the product of “Acceptor–Donor1–Donor2” plasmid could contain the attB-Bxb1 sequence (from “Donor2”). In the third round, such “Acceptor–Donor1–Donor2” plasmid could perform as the initial “Acceptor” plasmid to assemble with “Donor1”-like plasmid (Figure 3b and Figure S3a).
We also modified E. coli MG1655 strains that enable integrases to be constitutively expressed from the genome rather than “Helper” plasmids (Figure S4, this strategy could facilitate the plasmid assembly with efficiencies of more than 103 CFU/100 ng “Donor” plasmid). By doing this, a pair of selection marker (e.g., AmpR-ampicillin from “Helper” plasmids pLW1 and pLW2) could be released from the modified strains, enabling the “Donor3” plasmid construction and assembly (for the round 3 assembly between “Acceptor–Donor1–Donor2” and “Donor3”, Figure 3b). As expected, the assembled plasmids with one, two, or three “Donor” were linearized and showed the correct DNA sizes (Figure S3b). Furthermore, by utilizing three different fluorescent proteins, various E. coli cells with assembled plasmids exhibited the correct fluorescence phenotypes (Figure 3c and Figure S5).3.3. Assembled Plasmids Generate Colored E. coli
Following the demonstration of in vivo plasmid assembly, we sought to further expand this proof of concept to generate colored E. coli by matching different chromoprotein genes. Here, we integrated six different chromoprotein genes into the “Acceptor” plasmids for showing featured colors (Figure 4a). To enrich the diversity of color combination, we designed two “Donor” plasmids with different genetic circuits. “Donor1” consisted of an arabinose-induced chromoprotein expression circuit. Depending on the states of arabinose (with or without) and chromoprotein gene (six genes), “Donor1” could exhibit up to 12 (2 × 6) different state combinations. Similarly, “Donor2” contained rhamnose-induced chromoprotein expression circuit and could also perform up to 12 different states. In total, all the five elements could be orthogonally combined and theoretically showed up to 864 states in E. coli cells that harbor “Acceptor–Donor1–Donor2” plasmids (Figure 4b). To simply demonstrate the design, we built up two examples of “amajLime–CyOFP1–tsPurple” (Figure 4c,d) and “CyOFP1-amilCP-amajLime” (Figure S6). By following the colorimetric procedure, we successfully obtained different E. coli cell pellets with diverse colors. Furthermore, the standardization procedure was performed for these colors’ conversion from real cell colors to digital RGB colors. We envision that these colored E. coli cells might be further used to build living materials [23] and potentially served as living pigments for painting and education [24].

Figure 2. Serine integrase enables in vivo plasmid assembly. (a) Design of “Helper” plasmids (pLW1 and pLW2) that can respond to the arabinose induction to express serine integrases (int: integrase Bxb1 or phiC31). (b) Western-blot analysis of two serine integrases under the induction of 1% (w/v) arabinose (M: protein marker, NC: negative control without arabinose, T: total fraction, S: soluble fraction). (c) Plasmid assembly between “Acceptor” and “Donor” (attL: attachment site in the left, attR: attachment site in the right). (d) Schematic workflow of the assembled plasmid selection. (e) DNA fragment sizes of “Acceptor” (Ac, pLW3 for 2120 bp, pLW4 for 2115 bp), and “Acceptor-Donor” (Ac-D, pLW3-pLW5 for 4509 bp, pLW4-pLW6 for 4488 bp) plasmids after linearization by restriction endonuclease (M: DNA marker). (f) Efficiency of Bxb1-mediated assembly between “Acceptor” (pLW3) and “Donor” (pLW5). (g) Efficiency of phiC31-mediated assembly between “Acceptor” (pLW4) and “Donor” (pLW6). Each value (mean ± standard deviation) is calculated with three biological replicates and the error bar represents the standard deviation.

Figure 3. In vivo plasmid assembly cascade. (a) Design of three donors that contain fluorescent proteins (pLW9: mCherry, pLW10: sfGFP, pLW11: ECFP). After assembly, E. coli strains harboring these “Acceptor-Donor” plasmids (pLW3-pLW9, pLW3-pLW10, and pLW3-pLW11) were imaged under confocal microscope (scale bar: 10 µm). (b) Schematic workflow of in vivo plasmid assembly cascade. Note that the integrases of the first and second round were expressed from “Helper” plasmids (pLW1 and pLW2) in the wild type E. coli DH5α. The integrases of the third round were expressed from the modified E. coli MG1655 genome (see Figure S4). FP: fluorescent protein. (c) Assembly cascade workflow and fluorescence visualization of different E. coli strains that harbor assembled plasmids (scale bar: 10 µm). “Acceptor”: pLW3, “Donor1”: pLW9 with mCherry, “Donor2”: pLW12 with sfGFP, “Donor3”: pLW13 with ECFP.

Figure 4. Generation of colored E. coli by in vivo plasmid assembly. (a) Six basic colors were generated with six chromoproteins that are constitutively expressed from “Acceptor” plasmids (pLW25-pLW30). CP: chromoprotein. (b) Schematic design of three types of plasmids and theoretical combinations with five orthogonal elements. (c) Two color phenotypes were generated from the “Acceptor-Donor1” plasmid (pLW30-pLW31). Arabinose: 1% (w/v). (d) Four color phenotypes were generated from the “Acceptor-Donor1-Donor2” plasmid (pLW30-pLW31-pLW32). Arabinose: 1% (w/v), rhamnose: 1% (w/v).
4. Conclusions
Overall, we developed a serine integrase-based in vivo plasmid assembly approach. First, the assembly between “Acceptor” and “Donor” plasmid was successfully demonstrated. Second, a multi-round plasmid assembly cascade was established. Here, we showed three rounds of assembly and eventually constructed the “Acceptor–Donor1–Donor2–Donor3” plasmids that could constitutively express three different fluorescent proteins in one E. coli strain. Third, we expanded this approach to generate diverse chromoprotein-based plasmids and enable E. coli to show various colors. Looking forward, we anticipate that this in vivo plasmid assembly method together with currently various genetic tools will further accelerate DNA manipulation such as plasmid construction, genetic circuit design, and genome engineering. Moreover, the serine integrase-based tools will help advance the efforts to design, build, and test for fundamental and applied research in synthetic biology.
Supplementary Materials
The following supporting information can be found at: https://www.sciepublish.com/article/pii/99.
Author Contributions
Conceptualization, J.L. and F.B.; Methodology, L.W., Y.Z. and F.B.; Validation, L.W., F.B. and Y.Z.; Investigation, F.B. and L.W.; Resources, J.L.; Writing – Original Draft Preparation, F.B. and L.W.; Writing – Review & Editing, J.L., F.B. and W.-Q.L.; Supervision, J.L. and F.B.; Project Administration, J.L.; Funding Acquisition, J.L. and W.-Q.L.
Ethics Statement
Not applicable.
Informed Consent Statement
Not applicable.
Funding
This work was supported by grants from the National Natural Science Foundation of China (32171427 and 31971348).
Declaration of Competing Interest
The authors declare no conflicts of interest.
References
1.
Sheth RU, Wang HH. DNA-based memory devices for recording cellular events. Nat. Rev. Genet. 2018, 19, 718–732. [Google Scholar]
2.
Cameron DE, Bashor CJ, Collins JJ. A brief history of synthetic biology. Nat. Rev. Microbiol. 2014, 12, 381–390. [Google Scholar]
3.
Meng F, Ellis T. The second decade of synthetic biology: 2010–2020. Nat. Commun. 2020, 11, 5174. [Google Scholar]
4.
Grindley NDF, Whiteson KL, Rice PA. Mechanisms of site-specific recombination. Ann. Rev. Biochem. 2006, 75, 567–605. [Google Scholar]
5.
Ba F, Zhang Y, Wang L, Liu WQ, Li J. Applications of serine integrases in synthetic biology over the past decade. SynBio 2023, 1, 172–189. [Google Scholar]
6.
Merrick CA, Zhao J, Rosser SJ. Serine integrases: Advancing synthetic biology. ACS Synth. Biol. 2018, 7, 299–310. [Google Scholar]
7.
Stark WM. Making serine integrases work for us. Curr. Opin. Microbiol. 2017, 38, 130–136. [Google Scholar]
8.
Durrant MG, Fanton A, Tycko J, Hinks M, Chandrasekaran SS, Perry NT, et al. Systematic discovery of recombinases for efficient integration of large DNA sequences into the human genome. Nat. Biotechnol. 2023, 41, 488–499. [Google Scholar]
9.
Snoeck N, De Mol ML, Van Herpe D, Goormans A, Maryns I, Coussement P, et al. Serine integrase recombinational engineering (SIRE): A versatile toolbox for genome editing. Biotechnol. Bioeng. 2019, 116, 364–374. [Google Scholar]
10.
Elmore JR, Dexter GN, Baldino H, Huenemann JD, Francis R, Peabody V GL, et al. High-throughput genetic engineering of nonmodel and undomesticated bacteria via iterative site-specific genome integration. Sci. Adv. 2023, 9, eade1285. [Google Scholar]
11.
Ba F, Liu Y, Liu WQ, Tian X, Li J. SYMBIOSIS: synthetic manipulable biobricks via orthogonal serine integrase systems. Nucleic Acids Res. 2022, 50, 2973–2985. [Google Scholar]
12.
Siuti P, Yazbek J, Lu TK. Synthetic circuits integrating logic and memory in living cells. Nat. Biotechnol. 2013, 31, 448–452. [Google Scholar]
13.
Weinberg BH, Pham NTH, Caraballo LD, Lozanoski T, Engel A, Bhatia S, et al. Large-scale design of robust genetic circuits with multiple inputs and outputs for mammalian cells. Nat. Biotechnol. 2017, 35, 453–462. [Google Scholar]
14.
Roquet N, Soleimany AP, Ferris AC, Aaronson S, Lu TK. Synthetic recombinase-based state machines in living cells. Science 2016, 353, aad8559. [Google Scholar]
15.
Hsiao V, Hori Y, Rothemund PW, Murry RM. A population-based temporal logic gate for timing and recording chemical events. Mol. Syst. Biol. 2016, 12, 869. [Google Scholar]
16.
Yang L, Nielsen AAK, Fernandez-Rodriguez J, McClune CJ, Laub MT, Lu TK, et al. Permanent genetic memory with >1-byte capacity. Nat. Methods 2014, 11, 1261–1266. [Google Scholar]
17.
Abioye J, Lawson-Williams M, Lecanda A, Calhoon B, McQue AL, Colloms SD, et al. High fidelity one-pot DNA assembly using orthogonal serine integrases. Biotechnol. J. 2023, 18, 2200411. [Google Scholar]
18.
Gao H, Taylor G, Evans SK, Fogg PCM, Smith MCM. Application of serine integrases for secondary metabolite pathway assembly in Streptomyces. Synth. Syst. Biotechnol. 2020, 5, 111–119. [Google Scholar]
19.
Colloms SD, Merrick CA, Olorunniji FJ, Stark WM, Smith MCM, Osbourn A, et al. Rapid metabolic pathway assembly and modification using serine integrase site-specific recombination. Nucleic Acids Res. 2014, 42, e23. [Google Scholar]
20.
Ba F, Ji X, Huang S, Zhang Y, Liu WQ, Liu Y, et al. Engineering Escherichia coli to utilize erythritol as sole carbon source. Adv. Sci. 2023, 10, 2207008. [Google Scholar]
21.
Ba F, Zhang Y, Ji X, Liu WQ, Ling S, Li J. Expanding the toolbox of probiotic Escherichia coli Nissle 1917 for synthetic biology. Biotechnol. J. 2023, doi:10.1002/biot.202300327.
22.
Rakowski SA, Filutowicz M. Plasmid R6K replication control. Plasmid 2013, 69, 231–242. [Google Scholar]
23.
Peng R, Ba F, Li J, Cao J, Zhang R, Liu WQ, et al. Embedding living cells with a mechanically reinforced and functionally programmable hydrogel fiber platform. Adv. Mater. 2023, 35, 2305583. [Google Scholar]
24.
Jung JK, Rasor BJ, Rybnicky GA, Silverman AD, Standeven J, Kuhn R, et al. At-home, cell-free synthetic biology education modules for transcriptional regulation and environmental water quality monitoring. ACS Synth. Biol. 2023, 12, 2909–2921. [Google Scholar]