Found 4 results
Review
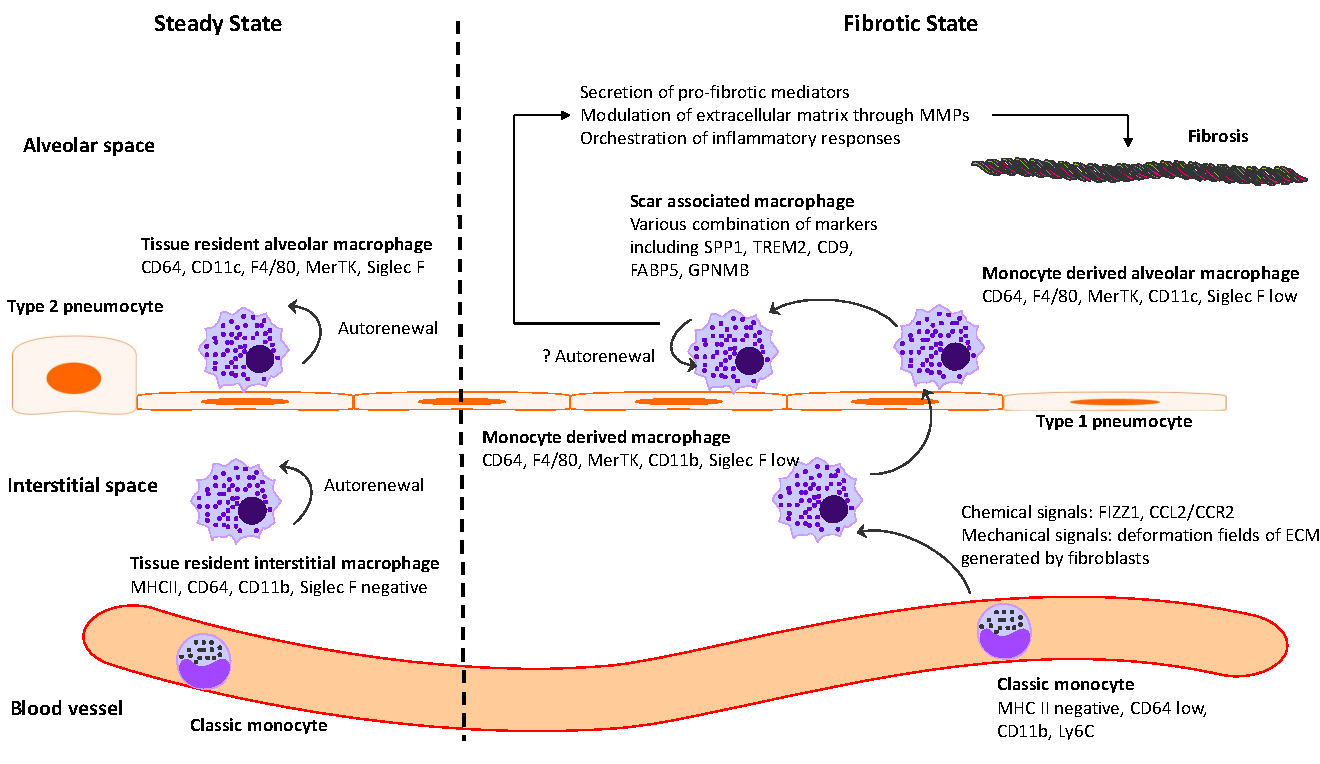
18 February 2025The Intersection between Immune System and Idiopathic Pulmonary Fibrosis—A Concise Review
Idiopathic pulmonary fibrosis (IPF) is marked by progressive alveolar destruction, impaired tissue regeneration, and relentless fibrogenesis, culminating in respiratory failure and death. A diverse array of resident and non-resident cells within the lung contribute to disease pathogenesis. Notably, immune cells, both resident and recruited, respond to cues from sites of lung injury by undergoing phenotypic transitions and producing a wide range of mediators that influence, initiate, or dictate the function, or dysfunction, of key effector cells in IPF pathology, such as alveolar epithelial cells, lung fibroblasts, and capillary endothelial cells. The role of the immune system in IPF has undergone an interesting evolution, oscillating from initial enthusiasm to skepticism, and now to a renewed focus. This shift reflects both the past failures of immune-targeting therapies for IPF and the unprecedented insights into immune cell heterogeneity provided by emerging technologies. In this article, we review the historical evolution of perspectives on the immune system’s role in IPF pathogenesis and examine the lessons learned from previous therapeutic failures targeting immune responses. We discuss the major immune cell types implicated in IPF progression, highlighting their phenotypic transitions and mechanisms of action. Finally, we identify key knowledge gaps and propose future directions for research on the immune system in IPF.
Review
28 October 2024Acute Exacerbations of Interstitial Lung Disease: Evolving Perspectives on Diagnosis and Management
Interstitial lung diseases (ILDs) are a heterogeneous group of chronic lung diseases caused by several potential etiologies but for many, the cause of a given ILD remains unknown. Accurate epidemiologic data are hard to find because of varying definitions, overlapping characteristics once thought to be unique to specific diseases, and ongoing changes in how ILDs are diagnosed and managed. In addition, there are significant variations in prevalence among different geographic populations, likely reflecting a combination of genetic and environmental differences. Certain risk factors, including exposure to cigarette smoke or environmental toxicants (asbestos, silica, fracking, coal dust, and air pollution), genetic mutations, and single nucleotide polymorphisms, have all been associated with developing interstitial lung disease. Due to the availability of high-resolution computed tomography (CT) scans, earlier and broader recognition of subtle imaging changes, and an aging worldwide population, the incidence and prevalence of ILDs are increasing. While a given cause of particular interstitial lung disease may vary, patients often experience breathlessness and a non-productive cough due to impaired alveolar gas exchange. Patients with ILD are prone to the development of acute exacerbations, marked by acute or chronic respiratory failure because of an acute exacerbation of the underlying lung disease. In this review, we discuss the definition of an acute exacerbation and comment on what is known about the underlying pathophysiology in exacerbations of idiopathic pulmonary fibrosis and other ILDs. We also emphasize the similarities in the clinical presentation of the acute exacerbations regardless of the underlying ILD, highlight key prognostic features of the diagnosis, and underscore the importance of interdisciplinary management of acute interstitial lung disease exacerbations.

Article
30 April 2024Arrestin beta 1 Regulates Alveolar Progenitor Renewal and Lung Fibrosis
The molecular mechanisms that regulate progressive pulmonary fibrosis remain poorly understood. Type 2 alveolar epithelial cells (AEC2s) function as adult stem cells in the lung. We previously showed that there is a loss of AEC2s and a failure of AEC2 renewal in the lungs of idiopathic pulmonary fibrosis (IPF) patients. We also reported that beta-arrestins are the key regulators of fibroblast invasion, and beta-arrestin 1 and 2 deficient mice exhibit decreased mortality, decreased matrix deposition, and increased lung function in bleomycin-induced lung fibrosis. However, the role of beta-arrestins in AEC2 regeneration is unclear. In this study, we investigated the role and mechanism of Arrestin beta 1 (ARRB1) in AEC2 renewal and in lung fibrosis. We used conventional deletion as well as cell type-specific deletion of ARRB1 in mice and found that Arrb1 deficiency in fibroblasts protects mice from lung fibrosis, and the knockout mice exhibit enhanced AEC2 regeneration in vivo, suggesting a role of fibroblast-derived ARRB1 in AEC2 renewal. We further found that Arrb1-deficient fibroblasts promotes AEC2 renewal in 3D organoid assays. Mechanistically, we found that CCL7 is among the top downregulated cytokines in Arrb1 deficient fibroblasts and CCL7 inhibits AEC2 regeneration in 3D organoid experiments. Therefore, fibroblast ARRB1 mediates AEC2 renewal, possibly by releasing chemokine CCL7, leading to fibrosis in the lung.

Article
28 November 2023Translational Studies Reveal the Divergent Effects of Simtuzumab Targeting LOXL2 in Idiopathic Pulmonary Fibrosis
The composition of extracellular matrix (ECM) is altered during pathologic scarring in damaged organs including the lung. One major change in the ECM involves the cross-linking of collagen, which promotes fibroblast to myofibroblast differentiation. We examined the role of lysyl oxidase (LOX)-like 2 in lung progenitors and fibroblasts cultured from normal or IPF lung samples and in a humanized mouse model of IPF using a monoclonal antibody (Simtuzumab). Primary lung fibroblasts from normal donor lungs and IPF lung explants were examined for expression of LOXL2. Targeting LOXL2 with Simtuzumab on normal and IPF fibroblasts was examined both in vitro and in vivo for synthetic, functional, and profibrotic properties. LOXL2 was increased at transcript and protein level in IPF compared with normal lung samples. In a dose-dependent manner, Simtuzumab enhanced differentiation of fibroblasts into myofibroblasts. Inhibition of LOXL2 also enhanced fibroblast invasion and accelerated the outgrowth of fibroblasts from dissociated human lung cell preparations. Finally, preventative or delayed delivery of Simtuzumab enhanced lung fibrosis in a humanized mouse model of pulmonary fibrosis. Consistent with its failure in a Phase 2 clinical trial, Simtuzumab exhibited no therapeutic efficacy in translational in vitro and in vivo assays.
