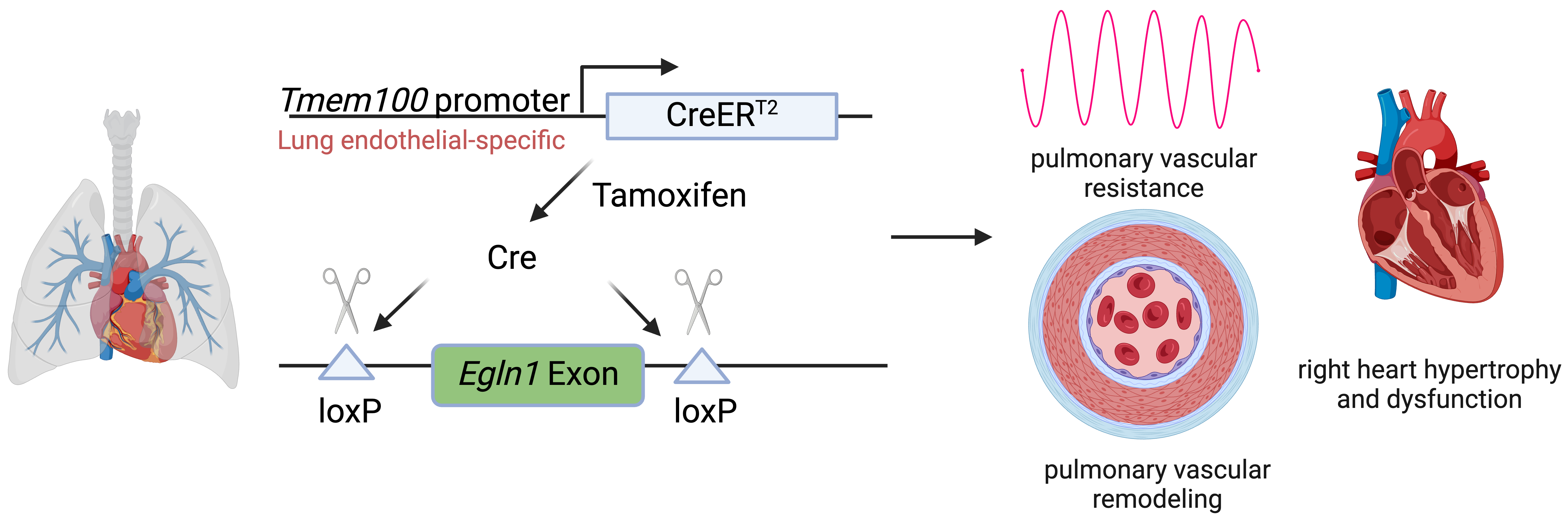
1. Introduction
Pulmonary hypertension (PH) is a devastating lung disease characterized by high blood pressure in the pulmonary arteries, which has the potential to lead to heart failure over time. Endothelial cell (EC) dysfunction plays a significant role in the development of PH [
1]. So far, many animal models have been established in mice to understand the role of key factors and genes in the ECs in the pathogenesis of PH. However, due to the ubiquitous expression of endothelial makers in all the organs, such as the pan-markers Cdh5 and Tie2, there are potential off-target effects in these pan-EC Cre mice models [
2]. The goal of this study is to demonstrate a lung endothelial-specific Cre tool for lung endothelial-specific manipulation in PH.
The lung possesses a unique morphology characterized by a thin layer of capillary ECs that facilitate efficient gas exchange, in contrast to other organs. Despite their critical importance in lung function, a Cre line specific to lung ECs has not been identified in the literature. Recent analysis using single-cell RNA sequencing of murine ECs across various organs revealed Tmem100 as one of the most enriched genes specifically in lung vasculature compared to other organ vasculatures [
3]. Our recent investigations employing immunostaining against TMEM100 in different human organs indicated high expression of TMEM100 exclusively in human lung ECs, with no detectable expression in ECs from other organs such as the heart, liver, and kidney. Our recent studies utilizing Tmem100-CreERT2; Ai6 mice illustrated predominant expression of Tmem100 (ZsGreen) in lung ECs of adult mice. Further analysis of Tmem100 (ZsGreen) expression in the lung demonstrated high expression in 92% of capillary ECs, 72% of arterial ECs, and 46% of venous ECs [
4].
We previously developed a new mouse model displaying severe PH through
Tie2Cre-mediated disruption of
Egln1, which encodes hypoxia-inducible factor (HIF) prolyl hydroxylase 2 (PHD2). This model, designated as
Egln1Tie2Cre, exhibits progressive obliterative vascular remodeling characterized by vascular occlusion and plexiform-like lesions, ultimately leading to right heart failure [
5]. We aimed to establish a lung endothelial-specific deletion of
Egln1 PH model using
Tmem100-CreERT2.
2. Methods and Materials
Tmem100-CreERT2;Ai6 mice were generated previously [
4].
Egln1f/f;Tmem100-CreERT2 (LiCKO) mice were generated by crossing the
Egln1flox/flox (WT) mice (JAX # 009672) and
Tmem100-CreERT2 mice (JAX # 014159).
Egln1f/f;Cdh5-CreERT2 (iCKO) mice were generated by crossing the
Egln1flox/flox (WT) mice and
Cdh5-CreERT2 mice (from Ralf Adams) [
6]. WT, LiCKO and iCKO mice aged 2–5 months were used in these studies. The animal care and study protocols were reviewed and approved by the Institutional Animal Care and Use Committees of the University of Arizona (#19-513).
2.1. Hemodynamic Measurement
Right ventricular systolic pressure (RVSP) was measured with a 1.4F pressure transducer catheter (Millar Instruments) and recorded with AcqKnowledge software (Biopac Systems Inc., Goleta, CA, USA) as described previously [
7,
8]. The catheter was inserted into the right ventricle in mice under anesthesia (100 mg ketamine/ 5mg xylazine /kg body weight, i.p.).
2.2. Echocardiography
Echocardiography was performed in the University of Arizona as described previously [
9]. Transthoracic echocardiography was performed using a VisualSonics Vevo 3100 ultrasound machine (FujiFilm VisualSonics Inc., Toronto, ON, Canada) with an MS550D (40 MHz) transducer. The right ventricle wall thickness during diastole (RV WTD) was obtained from the parasternal short-axis view at the papillary muscle level using M-mode. The RV cross-sectional area was obtained from the parasternal short-axis view at the papillary muscle level using B-mode. Pulmonary artery (PA) acceleration time and PA ejection time were obtained from the parasternal short-axis view at the aortic valve level using pulsed Doppler mode. The cardiac output (CO) was obtained from the parasternal short-axis view using M-mode.
2.3. Whole Organ Imaging, Immunofluorescent Staining, and Histological Staining
Various organs from
Tmem100-CreERT2;Ai6 mice post-tamoxifen treatment at approximately 2 weeks were perfused by PBS, followed by dissection and imaging under the fluorescent stereomicroscope. Mouse lung tissues were perfused with PBS, inflated with 50% OCT in PBS, and embedded in 100% OCT for cryosectioning. For immunofluorescent staining on these fresh frozen tissues, lung sections (5 μm) were fixed with 4% paraformaldehyde and blocked with 0.1% Triton X-100 and 5% normal goat serum at room temperature for 1 h. After 3 washes with PBS, the slides were incubated with anti-CD31 antibody (BD Bioscience, San Jose, CA, USA, Cat#550274, 1:25), anti-PHD2 (Cell Signaling Technology, Danvers, MA, USA, Cat#3293, 1:50) at 4 °C overnight, then incubated with Alexa 594 or 647-conjugated anti-rat or anti-rabbit IgG (Thermo Fisher Scientific, Waltham, MA, USA) at room temperature for 1 h. Nuclei were counterstained with DAPI mounting medium (SouthernBiotech, Birmingham, AL, USA).
For distal PA muscularization assessment, tissue slides were incubated with anti-α-SMA (Abcam, Cambridge, UK, Cat #ab5694, 1:300) at 4 °C overnight followed by Alexa 594 conjugated anti-rabbit IgG at room temperature for 1 h. Nuclei were counterstained with DAPI. Images were taken using Zeiss LSM710 confocal microscopy. α-SMA
+ vessels were quantified blindly in 40 fields per lung.
Mouse lung tissues were perfused with PBS and fixed with 10% formalin via tracheal instillation at a constant pressure (15 cm H
2O) and embedded in paraffin wax. Lung sections were stained with a Russel-Movat pentachrome staining kit (Cat #KTRMP, StatLab) according to the manufacturer’s protocol. For assessment of PA wall thickness, PAs from 40 images at 20X magnification were quantified blindly by Image J. Wall thickness was calculated by the distance between internal wall and external wall divided by the distance between external wall and the center of lumen.
2.4. Western Blot
Mice from various organ tissues were collected in RIPA lysis buffer supplemented with protease inhibitor cocktails (Sigma-Aldrich, St. Louis, USA). Approximately 20 μg of protein was loaded onto SDS-PAGE gels for Western Blotting. PVDF membranes were probed with anti-PHD2 (Cell Signaling Technology, Danvers, MA, USA, Cat #3293, 1:1000) or anti-β-actin (Sigma-Aldrich, St. Louis, MO, USA, Cat #A2228, 1:10,000) or GADPH (Sigma-Aldrich, St. Louis, MO, USA, Cat #G8795, 1:10,000) antibodies.
2.5. Statistical Analysis
Statistical analysis was performed using Prism 9 (Graphpad Software Inc., La Jolla, CA, USA). Statistical significance was determined by one-way ANOVA with Tukey’s post hoc analysis, calculating
p-values corrected for multiple comparisons. Two-group comparisons were analyzed using unpaired two-tailed
t-tests. A
p-value less than 0.05 indicated a statistically significant difference. All bar graphs represent mean ± SD.
3. Results
3.1. Tmem100-CreERT2 Mediates Lung Endothelial-Specific PHD2 Deletion
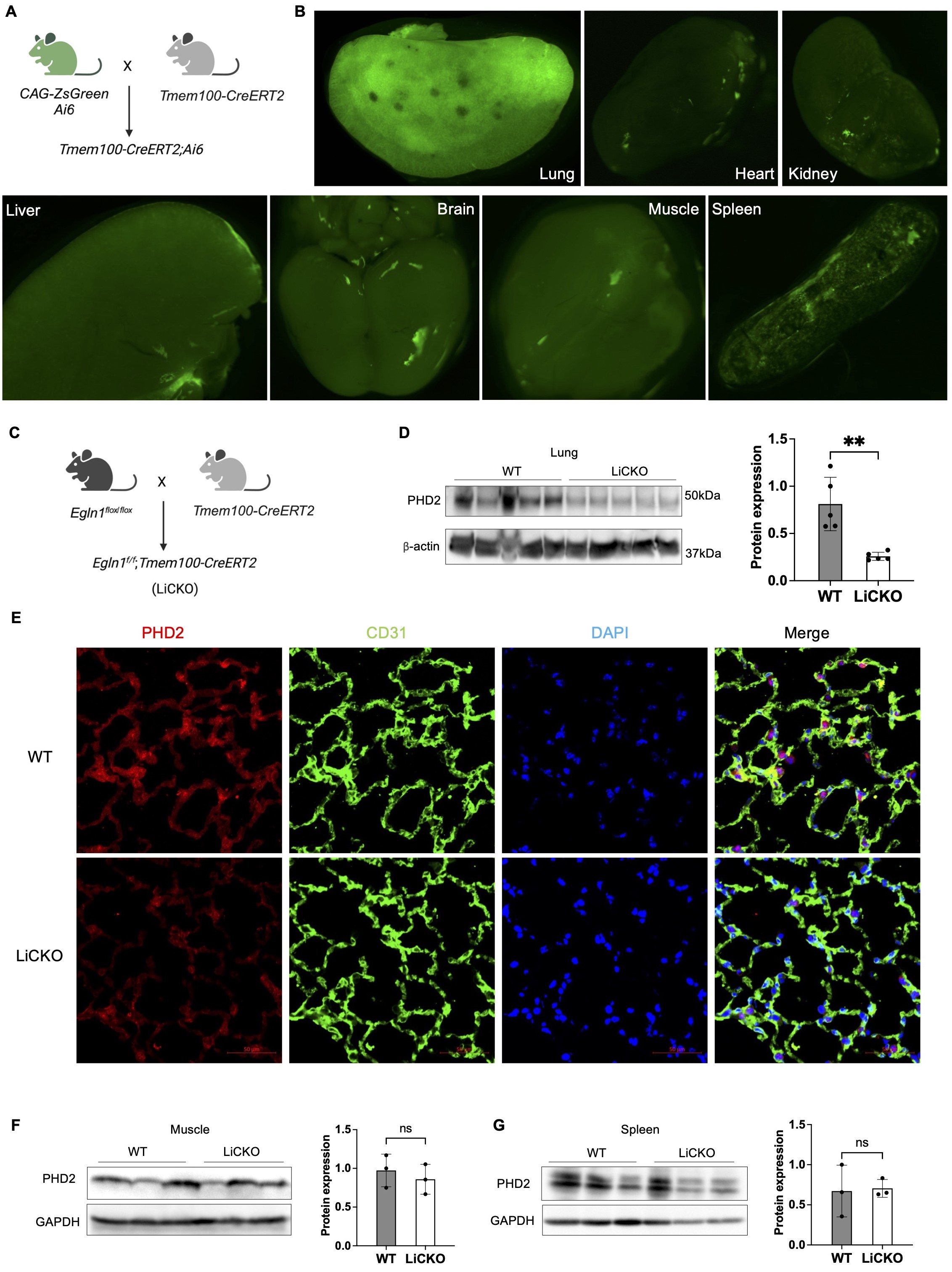
Employing single-cell RNA sequencing analysis and mice lineage tracing approaches, our previous studies demonstrated that Tmem100 is a gene specifically expressed in lung ECs.
Tmem100-CreERT2 mediated ZsGreen expression (
Tmem100-CreERT2;Ai6) in mice was mostly restricted to the lung vasculature compared to other organs, including the brain, heart, kidney and liver, as shown by immunofluorescent staining [
4]. In this study, we applied whole organ imaging using fluorescent stereoscopy on tissues isolated from
Tmem100-CreERT2;Ai6 mice treated with tamoxifen (A). We found that ZsGreen expression was highly present in the lung tissue and weakly observed in the heart, brain, kidney, spleen, muscle from isolated tissues (B).
To further determine whether
Tmem100-CreERT2 can mediate genetic deletion mainly in lung ECs in vivo, we crossbred Tmem100-CreERT2 mice with
Egln1flox/flox mice to generate
Egln1f/f;Tmem100-CreERT2 (LiCKO) mice (C). The LiCKO mice at the age of 7 weeks were injected with tamoxifen, and lung tissue was collected after one month. Western blotting analysis showed that lung PHD2 (encoded by
Egln1) was markedly reduced in the LiCKO lungs compared to WT lungs (D). Furthermore, immunostaining against PHD2 and the endothelial marker CD31 confirmed that PHD2 was reduced in the lung ECs of LiCKO compared to WT (E). Moreover, we also collected major organs from the mice, including the heart, spleen, and skeleton muscle for western blotting analysis. Our data showed no expression change in PHD2 in the heart, spleen, and skeleton muscle (F). Taken together,
Tmem100-CreERT2 mediated lung endothelial PHD2 deletion was restricted in the lung ECs.
. Lung endothelial-specific deletion of PHD2 in mice. (<b>A</b>) A diagram illustrating the strategy for generating lung endothelial-specific CreERT2 mice. (<b>B</b>) Whole organ imaging demonstrating the selective labeling of the lung by Tmem100-CreERT2 mice at the adult stage. ZsGreen indicates the presence of Tmem100 expression in organs. (<b>C</b>) A diagram illustrating the generation of lung endothelial-specific deletion of <i>Egln1</i> PH mouse model (<i>Egln1<sup>f/f</sup>;Tmem100CreERT2,</i> LiCKO). (<b>D</b>) Western blotting showed the knockdown of PHD2 expression in LiCKO lungs compared to WT (<i>Egln1<sup>flox/flox</sup></i>) lungs. (<b>E</b>) Immunostaining showing the knockdown of PHD2 in lung capillary ECs compared to WT lungs. Green signals indicate CD31<sup>+</sup> ECs. Red signals indicate PHD2 expression. (<b>F</b>) Western blotting showed that PHD2 expression was not affected in the skeletal muscle and spleen in LiCKO mice compared to WT mice. Student <i>t</i> test (<b>D</b>,<b>F</b>,<b>G</b>). <sup>∗∗</sup><i>p</i> < 0.01; ns, not significant. Scale bar: 50 μm (<b>E</b>).
Our previous studies and those of other groups have demonstrated that the loss of
Egln1 in ECs using
Tie2-Cre and
Cdh5-Cre or
Cdh5-CreERT2 mice induced spontaneous PH [
5,
10,
11,
12]. To determine whether LiCKO mice also develop PH, 7-week-old WT and LiCKO mice were challenged with tamoxifen for five consecutive days, followed by hemodynamic measurement at 1-month post-tamoxifen treatment. Hemodynamic data showed that the right ventricular systolic pressure (RVSP) was approximately 35 mmHg in LiCKO mice compared to 20 mmHg in WT mice (A). The ratio of RV to left ventricular plus septum (RV/LV+S) was also significantly increased up to 0.45 in LiCKO mice compared to 0.25 in WT mice (B). We then compared LiCKO mice with
Egln1f/f;Cdh5-CreERT2 (iCKO) mice after tamoxifen treatment and did not observe any difference in RVSP and RV/LV+S, suggesting that the efficacy of
Tmem100-CreERT2 is similar as
Cdh5-CreERT2 in inducing PH development. (C,D)
We then characterized the pulmonary vascular remodeling in LiCKO mice by measuring pulmonary wall thickness based on the Pentachrome staining and distal pulmonary arterial muscularization based on α-smooth muscle actin (α-SMA) staining. Our data showed significant increases in pulmonary wall thickness and distal muscularized pulmonary vessels (<50 μm) in LiCKO mice compared to WT mice (E,F). These data suggest that
Tmem100-CreERT2 mediated
Egln1 deletion induces spontaneous PH in mice.
Patients with PAH largely die due to right heart failure [
13]. We next performed echocardiography measurements to determine whether LiCKO mice develop right heart dysfunction. Our echocardiography measurements showed that the LiCKO exhibited significant increases in RV wall thickness during diastole (RV WTD) (G), while a decrease in PA function, assessed by PA acceleration time/ejection time (AT/ET) ratio (H), and the right ventricular fractional area changes (RV FAC), indicative of RV contractility (I). We did not observe any difference in cardiac output (CO) and heart rate (HR) between LiCKO mice and WT mice (J,K). Taken together, these data suggest that
Tmem100-CreERT2 mediated
Egln1 deletion induces spontaneous PH and right heart dysfunction in mice.
. Lung endothelial-specific deletion of PHD2 induced PH and right heart dysfunction in mice. (<b>A</b>) Hemodynamic measurements showed that LiCKO mice developed elevated RVSP compared to WT mice after tamoxifen treatment. (<b>B</b>) Cardiac dissection revealed an increase in the RV/LV+S ratio, indicative of right heart hypertrophy, in LiCKO mice. (<b>C</b>,<b>D</b>) LiCKO mice developed similar degree of PH phenotypes compared to iCKO mice (<i>Egln1<sup>f/f</sup>;Cdh5-CreERT2</i>). (<b>E</b>,<b>F</b>) Representative images from Russel-Movat Pentachrome staining and quantification of PA wall thickness demonstrated an increase in pulmonary vascular remodeling in LiCKO lungs compared to WT lungs. (<b>G</b>,<b>H</b>) Exemplary images and quantification data showed an increase in the muscularization of distal pulmonary vessels in LiCKO mice. Lung sections were subjected to immunostaining using anti-smooth muscle α-actin (SMA). (<b>I</b>) Echocardiography measurements revealed an increase in RV wall thickness during diastole (RV WTD) in LiCKO mice compared with WT mice. (<b>J</b>) Impaired RV fraction area change (RV FAC), indicating enhanced RV contractility, in LiCKO mice compared with WT mice. (<b>K</b>) An increased ratio of pulmonary artery acceleration time to ejection time (PA AT/ET) in LiCKO mice compared with WT mice. (<b>L</b>,<b>M</b>) There is no significant change of heart rate (HR), cardiac output (CO) between WT and LiCKO mice. Student <i>t</i> test (<b>A–D</b>, <b>I–M</b>). <sup>∗∗∗</sup><i>p</i> < 0.001; <sup>∗∗∗∗</sup><i>p</i> < 0.0001; ns, not significant. Scale bar: 50 μm (<b>G</b>).
4. Discussion
The present studies demonstrated that
Tmem100-CreERT2 mediated deletion of
Egln1 specifically causes the decrease of PHD2 in lung ECs but not in other organs. Loss of
Egln1 in lung ECs leads to spontaneous PH and right heart dysfunction.
Tie2-Cre and
Cdh5-Cre/CreERT2 are the wildly used pan-EC mouse models. However, there are potential off-target effects of these pan-EC mouse models, which restrict their use in lung vascular research. For example, pan-endothelial deletion of β-catenin in mice (
Ctnnb1f/f;EndoSCL-CreERT2) led to blood-brain barrier leakage and lethality shortly after gene deletion induced by tamoxifen [
14], preventing the utilization of this model in lung vasculature studies. Our study demonstrated that Tmem100-CreERT2 mediated PHD2 deletion is restricted to the lung but not to other organs. We also compared the efficacy of
Tmem100-CreERT2 with
Cdh5-CreERT2 mice and showed the similar effects in inducing PH development.
In the adult stage, Tmem100 exhibits its highest expression levels in the lung, with comparatively lower expression in the brain, heart, and muscle, as revealed by northern blot analysis [
15]. Additional investigations have revealed its significant presence in the bone, esophagus, PaS cells, and proliferating zone chondrocytes through the utilization of reporter mice [
16,
17]. Our recent exploration, utilizing Tmem100-CreERT2; Ai6 mice, has unveiled predominant expression of Tmem100 (ZsGreen) in lung ECs, the right atrium, and cardiac valves in adult mice [
4,
18]. However, existing Tmem100-related mouse models have limitations as they were solely investigated in specific organs or developmental stages to assess Tmem100 expression. There remains a possibility that Tmem100 expression or depletion occurs in a spatiotemporal fashion within particular cells.
Accumulating evidence shows that PH is a systemic metabolic disease [
19,
20,
21]. PH primarily leads to right-sided heart failure, triggering a multifaceted clinical syndrome that impacts various organ systems such as the left heart, brain, kidneys, liver, gastrointestinal tract, skeletal muscle, as well as the endocrine, immune, and autonomic systems. Understanding the interorgan crosstalk and interdependent mechanisms will help reduce the mortality and morbidity in patients.
Thus,
Tmem100-CreERT2 mouse model might be an ideal tool for dissecting the role of lung-specific ECs without affecting vascular beds in organs outside the lung. LiCKO mice primarily affect lung vasculature and PH development, which might be a novel model to investigate interorgan crosstalk in PH pathogenesis.
Acknowledgments
We thank Dr. Ralf Adams (Max Planck Institute) for sharing the Cdh5-CreERT2 mice and Dr. You-Yang Zhao (Northwestern University) for sharing the Egln1f/f mice.
Author Contributions
B.L. and Z.D. conceived the experiments and interpreted the data. B.L., D.Y., X.M., H.Z., K.R., X.X. and Z.D. designed, performed experiments, and analyzed the data. V.V.K., M.B.F. and S.Q. provided the resources, B.L. wrote the manuscript. Z.D. revised the manuscript.
Ethics Statement
Not applicable.
Informed Consent Statement
Not applicable.
Funding
This work was supported in part by NIH grant R00HL138278, R01HL158596, R01HL62794, R01HL169509, R01HL170096, AHA Career Development Award 20CDA35310084, The Cardiovascular Research and Education Foundation, Arizona Biomedical Research Centre funding (RFGA2022-01-06), and University of Arizona institution funding to Z.D.
Declaration of competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
1.
Evans CE, Cober ND, Dai Z, Stewart DJ, Zhao Y-Y. Endothelial cells in the pathogenesis of pulmonary arterial hypertension.
Eur. Respir. J. 2021,
58, 2003957.
[Google Scholar]
2.
Payne S, Val SD, Neal A. Endothelial-specific cre mouse models is your cre CREdibile?
Arter. Thromb. Vasc. Biol. 2018,
38, 2550–2561.
[Google Scholar]
3.
Kalucka J, de Rooij LPMH, Goveia J, Rohlenova K, Dumas SJ, Meta E, et al. Single-Cell Transcriptome Atlas of Murine Endothelial Cells.
Cell 2020,
180, 764–779.
[Google Scholar]
4.
Liu B, Yi D, Yu Z, Pan J, Ramirez K, Li S, et al. TMEM100, a Lung-Specific Endothelium Gene.
Arter. Thromb. Vasc. Biol. 2022,
42, 1495–1497.
[Google Scholar]
5.
Dai Z, Li M, Wharton J, Zhu MM, Zhao YY. Prolyl-4 Hydroxylase 2 (PHD2) deficiency in endothelial cells and hematopoietic cells induces obliterative vascular remodeling and severe pulmonary arterial hypertension in mice and humans through hypoxia-inducible factor-2α.
Circulation 2016,
133, 2447–2458.
[Google Scholar]
6.
Sörensen I, Adams RH, Gossler A. DLL1-mediated Notch activation regulates endothelial identity in mouse fetal arteries.
Blood 2009,
113, 5680–5688.
[Google Scholar]
7.
Liu B, Peng Y, Yi D, Machireddy N, Dong D, Ramirez K, et al. Endothelial PHD2 deficiency induces nitrative stress via suppression of caveolin-1 in pulmonary hypertension.
Eur. Respir. J. 2022,
60, 2102643.
[Google Scholar]
8.
Liu B, Yi D, Pan J, Dai J, Zhu MM, Zhao Y-Y, et al. Suppression of BMP signaling by PHD2 deficiency in Pulmonary Arterial hypertension.
Pulm. Circ. 2022,
12, e12056.
[Google Scholar]
9.
Yi D, Liu B, Ding H, Li S, Li R, Pan J, et al. E2F1 Mediates SOX17 Deficiency–Induced Pulmonary Hypertension.
Hypertension 2023,
80, 2357-2371.
[Google Scholar]
10.
Kapitsinou PP, Rajendran G, Astleford L, Michael M, Schonfeld MP, Fields T, et al. The endothelial PHD2/HIF-2 axis regulates pulmonary artery pressure in mice.
Mol. Cell. Biol. 2016,
64, 961–962.
[Google Scholar]
11.
Wang S, Zeng H, Xie X-J, Tao Y-K, He X, Roman RJ, et al. Loss of prolyl hydroxylase domain protein 2 in vascular endothelium increases pericyte coverage and promotes pulmonary arterial remodeling.
Oncotarget 2016,
7, 58848–58861.
[Google Scholar]
12.
Elamaa H, Kaakinen M, Nätynki M, Szabo Z, Ronkainen V-P, Äijälä V, et al. PHD2 deletion in endothelial or arterial smooth muscle cells reveals vascular cell type-specific responses in pulmonary hypertension and fibrosis.
Angiogenesis 2022,
25, 259–274.
[Google Scholar]
13.
Mocumbi A, Humbert M, Saxena A, Jing Z-C, Sliwa K, Thienemann F, et al. Pulmonary hypertension.
Nat. Rev. Dis. Primers. 2024,
10, 5.
[Google Scholar]
14.
Tran KA, Zhang X, Predescu D, Huang X, Machado RF, Göthert JR, et al. Endothelial β-Catenin Signaling Is Required for Maintaining Adult Blood–Brain Barrier Integrity and Central Nervous System Homeostasis.
Circulation 2016,
133, 177–186.
[Google Scholar]
15.
Somekawa S, Imagawa K, Hayashi H, Sakabe M, Ioka T, Sato GE, et al. Tmem100, an ALK1 receptor signaling-dependent gene essential for arterial endothelium differentiation and vascular morphogenesis.
Proc. Nat. Acad. Sci. USA 2012,
109, 12064–12069.
[Google Scholar]
16.
Kinugasa-Katayama Y, Watanabe Y, Hisamitsu T, Arima Y, Liu NM, Tomimatsu A, et al. Tmem100-BAC-EGFP mice to selectively mark and purify embryonic endothelial cells of large caliber arteries in mid-gestational vascular formation.
Genesis 2021,
59, 1–11.
[Google Scholar]
17.
Vesprey A, Suh ES, Göz Aytürk D, Yang X, Rogers M, Sosa B, et al. Tmem100- and Acta2-Lineage Cells Contribute to Implant Osseointegration in a Mouse Model.
J. Bone Miner. Res. 2021,
36, 1000–1011.
[Google Scholar]
18.
Pan J, Liu B, Dai Z. The Role of a Lung Vascular Endothelium Enriched Gene TMEM100.
Biomedicines 2023,
11, 937.
[Google Scholar]
19.
Paulin R, Michelakis ED. The metabolic theory of pulmonary arterial hypertension.
Circul. Res. 2014,
115, 148–164.
[Google Scholar]
20.
Nickel NP, Yuan K, Dorfmuller P, Provencher S, Lai YC, Bonnet S, et al. Beyond the lungs: Systemic manifestations of pulmonary arterial hypertension.
Am. J. Res. Crit. Care Med. 2020,
201, 148–157.
[Google Scholar]
21.
Zamanian RT, Hansmann G, Snook S, Lilienfeld D, Rappaport KM, Reaven GM, et al. Insulin resistance in pulmonary arterial hypertension.
Eur. Res. J. 2009,
33, 318–324.
[Google Scholar]
 Dan Yi
1,2,3
Dan Yi
1,2,3