The alveolar epithelium of the lung comprises AT1 cells and AT2 cells. As alveolar stem cells, AT2 cells are capable of self-renewal and differentiation into AT1 cells during homeostasis and repair [
1]. Identifying the cellular origins of AT2 cells following lung damage is vital for uncovering new therapeutic targets for various lung diseases to enhance tissue regeneration. However, the sources of AT2 cells have been subject to debate due to potential non-specific labeling by traditional genetic lineage tracing methods. Site-specific DNA recombination systems like the Cre-loxP system are frequently employed in cell lineage studies. The accuracy of promoter-driven Cre expression is critical for ensuring cell-specific labeling. If Cre recombinase is expressed under a promoter active in the target cells but also in unintended or "unwanted" cell types, then both cell types will be labeled following Cre-loxP recombination. This co-labeling can result in misleading conclusions, as it suggests a lineage relationship where none may exist. It is crucial to ensure the specificity of promoter activity to avoid such inaccuracies in cell fate-mapping studies [
2]. Therefore, it is essential to validate the specificity of lineage tracing tools prior to their use in cell fate-mapping studies.
Recent lineage tracing studies have indicated that Hopx-CreER-labeled AT1 cells [
3,
4,
5,
6,
7] and
Scgb1a1-CreER-labeled club cells [
8,
9] are progenitors of AT2 cells following alveolar damage. However, the labeling specificity of these genetic tools has not been rigorously evaluated. In our recent study, “Tracing the origin of alveolar stem cells in lung repair and regeneration” [
10], we undertook a detailed examination of the non-specific labeling associated with conventional
Hopx-CreER and
Scgb1a1-CreER tools. Moreover, we employed dual recombinase-mediated genetic strategies to re-examine the cellular origin of AT2 cells during lung repair, enhancing the accuracy of lineage tracing in this context ().
Our study uncovered that the
Hopx-CreER genetic tool could inappropriately label unintended or “unwanted” cell types, including club cells, AT2 cells, bronchioalveolar stem cells (BASCs), and ciliated cells, besides the intended AT1 cells. Both AT2 cells and BASCs have been previously identified as stem/progenitor cells for regenerating AT2 cells post-lung injury [
11,
12,
13,
14,
15,
16,
17]. Similarly, another tool,
Ager-CreER, also targeted AT2 cells and a subset of BASCs, in addition to AT1 cells. Likewise, the
Scgb1a1-CreER tool could ectopically label BASCs and a subset of AT2 cells, in addition to its intended target, club cells [
18]. Currently, there are no such specific Cre tools available that completely avoid this issue of non-specific labeling.
To address this, we utilized the Dre-rox system, another homologous recombinase [
19], in conjunction with the Cre-loxP system. By exploiting the mutual exclusion of ectopic labeling between
Ager-CreER and
Hopx-2A-DreER, we achieved an 83% efficiency in labeling Ager
+Hopx
+ AT1 cells using an intersectional dual genetic reporter system (
R26-RSR-LSL-tdT). Additionally, we employed another dual recombination system (R26-NR2), which specifically traced Hopx
+ AT1 cells with approximately 98% efficiency. Fate-mapping analysis confirmed that AT1 cells are terminally differentiated and do not de-differentiate into AT2 cells during injuries caused by pneumonectomy (PNX), bleomycin, or hyperoxia. These findings align with recent studies indicating that Hopx
+Igfbp2
+ AT1 cell subpopulations do not de-differentiate into AT2 cells in PNX-injured lungs [
20]. The potential for AT1 cells to convert to AT2 cells under extreme conditions remains an area for further investigation. Such a transformation might require a stronger impetus, likely through genetic manipulation, to trigger a de-differentiation program in these terminally differentiated cells [
21,
22]. Exploring these conditions could open new pathways for enhancing lung repair and regeneration, offering promising avenues for therapeutic interventions in lung diseases.
For club cells, AT2 cells, and BASCs, we employed an alternative intersectional dual genetic system (
R26-TLR), which allowed for the simultaneous and specific labeling of these three distinct cell populations within a single mouse. This approach facilitated precise elucidation of their contributions to AT2 cell regeneration following lung injury. Utilizing various injury models revealed that the cellular plasticity of these cells varied depending on the type of lung injury encountered. In the PNX injury model, the regeneration of AT2 cells predominantly occurred through self-renewal, with minimal contribution from club cells and BASCs. However, in the bleomycin-induced injury model, both club cells and BASCs significantly contributed to the regeneration of a substantial portion of AT2 and AT1 cells.
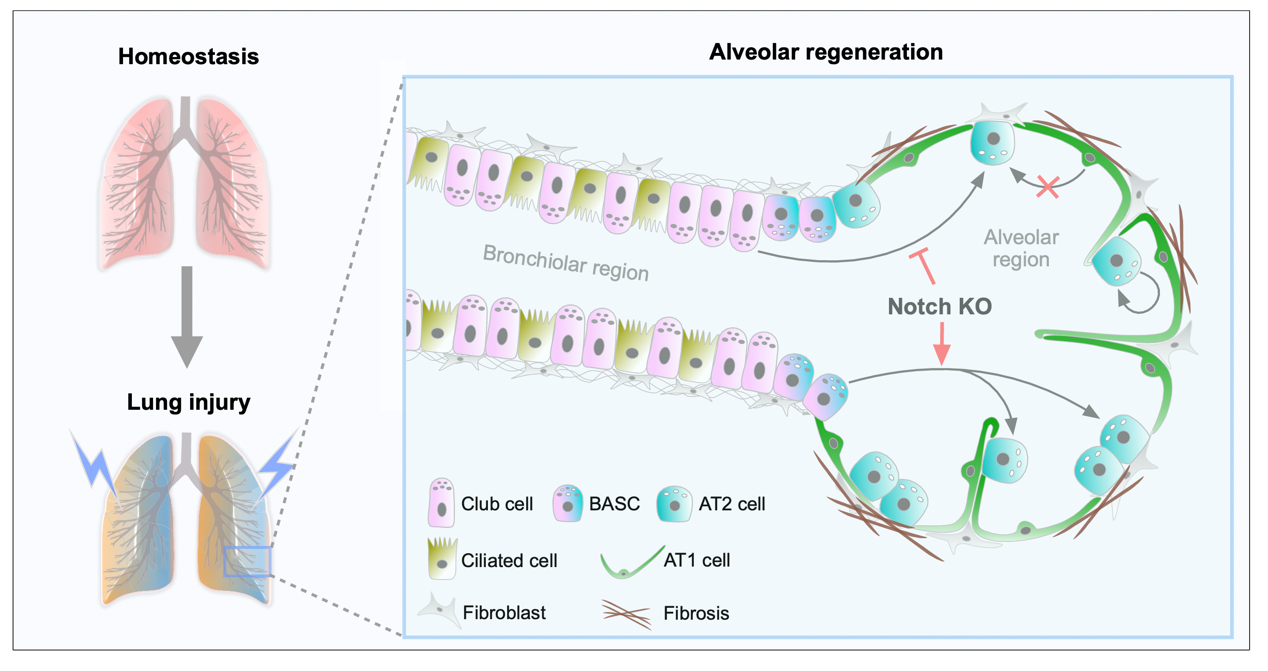
To further explore the regenerative potential of club cells, we constructed two severe alveolar injury models. In the first model, we impaired the regenerative capacities of AT2 cells and bronchioalveolar stem cells (BASCs) in a bleomycin-injury scenario by ectopically expressing p21. In the second, we induced the depletion of AT2 cells and BASCs through diphtheria toxin (DT)-mediated cell death. Remarkably, club cells exhibited substantial plasticity and regenerative potential, contributing to the reconstitution of the majority of AT2 cells in some lung lobes following severe alveolar injury. This finding suggests that when the alveolar epithelium's native repair mechanisms are inhibited, club cells in the airway region are capable of completely reconstructing the alveolar structures in certain damaged areas. Employing combined single-cell RNA sequencing (scRNA-seq), cell-specific gene knockouts, and multiple cell lineage tracing techniques, we identified that Notch signaling has divergent regulatory effects on the differentiation of club cells and BASCs into AT2 cells post-injury. Specifically, inhibition of Notch signaling enhanced the conversion of BASCs into AT2 cells, whereas its activation prevented this transformation. Conversely, inhibition of Notch signaling limited the differentiation of club cells into AT2 cells, instead favoring their development into ciliated cells.
In pulmonary diseases such as chronic obstructive pulmonary disease, idiopathic pulmonary fibrosis, and lung diseases induced by smoking, the damage to or senescence of AT2 cells significantly impairs the repair and regeneration of lung epithelial tissues. The transplantation of lung progenitor or stem cells presents a critical therapeutic strategy for treating these diseases [
23]. Given the remarkable plasticity of club cells and BASCs in alveolar regeneration, directly delivering these progenitor cells into the alveolar region might facilitate immediate cell fate conversion, potentially enhancing the effectiveness of cell-based therapies. Furthermore, pharmacologically targeting pathways such as Notch signaling to modulate this plasticity offers an additional promising approach to develop successful treatments for pulmonary diseases.
It should be noted that our findings appear to contradict a recent study, which suggested that inhibition of Notch signaling facilitates the transition of club cells to AT2 cells during repair processes [
9]. This discrepancy may arise because the previous study employed
Scgb1a1-CreER to trace both club cells and BASCs, complicating the differentiation of the effects of Notch signaling in each cell type [
9]. Moreover, another study identified a distinct population of club cells, termed respiratory airway secretory cells (RAS), located in the human distal airways [
24]. These cells exhibited a hybrid expression pattern characteristic of both club and AT2 cells, suggesting their role as progenitors in AT2 cell regeneration in pulmonary diseases [
24]. In vitro experiments demonstrated that inhibition of Notch signaling promotes the differentiation of RAS into AT2 cells [
24], aligning with our findings concerning the regulation of BASCs by Notch. Given the similar features between BASCs and RAS, BASCs may provide an ideal in vivo model for studying the molecular and cellular mechanisms involved in lung repair.
In conclusion, by employing dual recombinases-mediated genetic tracing, we have precisely labeled multiple types of pulmonary epithelial cells and revealed their cell capacity for contributing to AT2 cell regeneration following lung injury. The dual genetic strategies developed in this study surpass the conventional Cre-loxP system in targeting specificity and, in conjunction with cell lineage tracing, facilitate a more accurate investigation of molecular mechanisms. This advanced genetic lineage tracing technique will offer significant potential for exploring cell plasticity and fate decisions in various organs across development, disease states, and regenerative processes.
. The cellular origin of AT2 stem cells is diverse after lung injury. In the bleomycin-induced alveolar injury model, the regenerated AT2 cells originate from multiple sources, including club cells, BASCs, and the self-renewal of existing AT2 cells, but not from AT1 cells. Furthermore, Notch signaling exhibits opposite regulatory functions in the differentiation of club cells and BASCs into AT2 cells post-injury. Specifically, inhibition of Notch signaling decreases the conversion of club cells into AT2 cells, whereas promoting BASCs differentiation into AT2 cells.
We thank the Shanghai Model Organisms Center for mouse generation. We also thank Kathy O. Lui for valuable suggestions on this study, Hongkui Zeng for sharing the R26-tdT and R26-RSR-LSL-tdT mice, and Hongbin Ji for sharing the Sftpc-DreER mice.
K.L. and B.Z. designed the study and wrote the paper.
All mouse experiments in this study were strictly performed within the committee’s guidelines of the Institutional Animal Care and Use Committee (IACUC) of the Center for Excellence in Molecular Cell Science, Chinese Academy of Sciences.
Not applicable.
This study was supported by the National Natural Science Foundation of China (82088101, 32370897, 32100648), the National Key Research & Development Program of China (2023YFA1800700), and the New Cornerstone Science Foundation through the New Cornerstone Investigator Program and the XPLORER PRIZE.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.