1. Introduction
Asthma is a heterogeneous disease characterized by chronic airway inflammation, airway hyperresponsiveness, and reversible airflow obstruction, which affects about 262 million worldwide and causes an economic burden of $80 billion in the United States alone [
1,
2]. The etiology of asthma is complex and far from completely understood, with genetic, immunological, and environmental factors all contributing to the development of asthma [
3,
4,
5,
6]. In recent years, the human microbiome has proven to have had a significant impact on the development of the host immune system and overall maintenance of health, offering an excellent opportunity to deepen our understanding of the mechanisms of asthma from a new perspective [
7,
8].
The human nasopharynx is colonized by a diverse community of commensal microbiota, which serve as “gatekeepers” for respiratory health [
9]. Though limited evidence is available to date, the role of nasal microbiome in asthma has garnered increasing attention because of its inherent connection and shared biology with upper and lower airways, and paramount exposure to pathogens and bacteria. An early-life dysbiosis in the microbiota can often lead to an increased risk of developing asthma [
10,
11,
12,
13]. Based on a prospective design, McCauley et al. performed an amplicon sequencing targeting the 16S ribosomal RNA gene. They found that children’s nasal microbiome exhibited seasonal variability, which could be associated with an increased risk of asthma exacerbation [
14]. However, unrobust, and even contradictory results still exist in previous studies, which may be partly due to their relatively small sample sizes. For example, Zhou et al. examined in 102 samples the association of nasal microbial diversity with the yellow zone, a period of early loss of the control of asthma, and found a significantly higher Shannon’s Index in yellow zone samples. Another study of 68 nasal brushing samples reported a decreased α diversity in asthmatics compared to healthy control samples [
15,
16]. Interestingly, a recent systematic review showed that, among ten studies on respiratory microbiota and asthma included, only four reported a positive association between α diversity and asthma risk, while two showed a negative association and the other four showed no significance. A meta-analysis of all 10 studies showed no significance in the differences between Shannon, Chao1, and Simpsons index [
17]. In addition, using amplicon sequencing, most studies usually stopped at reporting associations at the genus or even higher levels [
4,
14,
18]. These limitations greatly obstructed the understanding of asthma pathogenesis from the perspective of the nasal commercial microbiome and its application in management and treatment.
To address these limitations, in the current study, we utilized two published RNA-seq datasets with relatively large sample sizes, employing meta-transcriptomic analytical strategies to compare the nasal microbiome composition between asthmatic and healthy control individuals through diversity analysis and meta-analysis of two independent differential abundance analyses. This project aims to determine differences in microbial diversity between asthma and non-asthma patients and facilitate further research into an understudied yet insightful portion of the human microbiome.
2. Materials and Methods
2.1. Data Selection and Preprocessing
We analyzed data from two independent multicentered studies on the genetic factors influencing asthma. The GALA II study enrolls 4427 children of Puerto Rican heritage aged 8–21, chosen from 10 sites across both the United States and Puerto Rico [
19]. NA-seq data was gathered by taking nasal airway epithelium brushings of 694 participants; 441 were characterized as “asthmatic” and 253 were characterized as “healthy control”. The second dataset is from the Consortium on Asthma among African-ancestry Populations in the Americas (CAAPA) study [
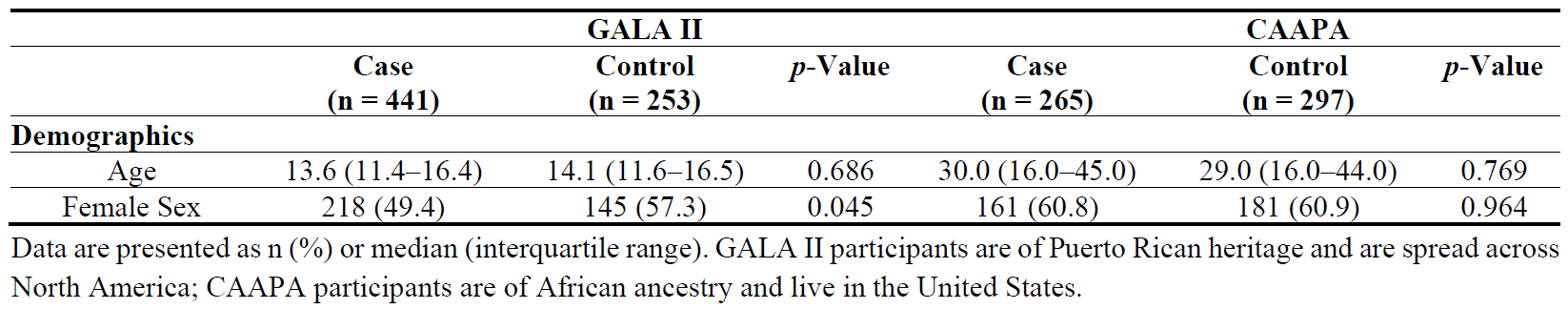
20], which enrolled 14,548 individuals of all ages of African Ancestry across 15 locations. Nasal airway epithelium brushings were taken from 562 participants, 265 “asthmatic” individuals and 297 “not asthmatic” individuals ().
. Baseline characteristics of patients in GALA II and CAAPA study datasets.
The raw FASTQ files from both studies (GSE152004 and GSE240567) were downloaded from the GEO platform. We then utilized the nf-core [
21] tax-profiler [
22] pipeline to preprocess the FASTQ files and extract the microbiome components from the samples. First, RNA-seq samples underwent quality control using fastp [
23]. The sequencing data were then aligned to the human reference genome (GRCh38 [
24]) with Bowtie [
25] to remove the host genome components. The remaining reads were annotated and quantified using Kraken2 [
26] with the Maxikraken2_1903_140GB database (March 2019), which includes archaea, bacteria, fungi, protozoa, and viral sequences from the Loman Lab [
27]. Finally, we generated both an absolute abundance matrix and a relative abundance matrix for all 694 samples using Bracken for downstream analysis. Asthma status and sequencing run IDs (SRR) in the abundance matrices were matched to the samples using accession numbers (GSM) from the series matrix file as a reference.
2.2. Diversity Analysis
Using the derived absolute abundance matrix and relative abundance matrix, we performed α and β diversity analysis to evaluate the similarity and differences in nasal microbial community composition between asthmatic and non-asthmatic samples in each independent dataset. Four α diversity indices (Shannon, Berger-Parker, Inverse Simpsons, and Fishers), which quantify the species richness within a single sample, were calculated for each sample using KrakenTools [
17,
28]. A Wilcoxon rank-sum test was conducted to evaluate differences between groups. An increase in the index value indicates greater α diversity for Shannon, Inverse Simpsons, and Fishers indices. In contrast, a lower value corresponds to greater α diversity for the Berger-Parker index. For β diversity analysis, which is a comparison of microbial diversity between two groups of samples, a Bray-Curtis dissimilarity matrix using KrakenTools was first calculated, followed by a permutational multivariate analysis of variance (PERMANOVA) using the vegan (version 2.6.6.1) package for differences comparison [
29]. Principal Coordinate Analysis (PcoA) was also employed to visualize the distance across samples based on the calculated dissimilarity matrix. Results with
p-value < 0.05 were defined as statistically significant.
2.3. Differential Abundance Analysis
Species with a mean abundance of less than 2 and a median abundance of less than 2 across all samples were removed from the absolute abundance matrix. Then, using the DESeq2 package [
30], differential abundance analysis (DAA) was first conducted separately on each independent dataset to identify taxa associated with asthma. We controlled for age and sex as covariates in the differential abundance analyses of both GALA II and CAAPA study datasets. Differentially abundant taxa (DAT) were defined as microbial taxa with Benjamini & Hochberg adjusted
p-value < 0.05. Finally, a fixed-effects meta-analysis was conducted using the metafor package [
31] (version 4.6.0) to provide a more precise estimate of DAT across studies. Data preprocessing was performed using KrakenTools in Python (version 3.9.13), and all statistical analyses were conducted in R (version 4.4.1).
3. Results
The baseline characteristics of the participants in both the GALA II and CAAPA study are detailed in . The GALA II study had 441 cases and 253 control samples. The median age of the case group was 13.6 years, while the control group had a mean age of 14.1 years. The proportion of females was significantly higher in the control group (57.3%) compared to the case group (49.4%) (
p < 0.05). The CAAPA study included 265 cases and 297 controls, with mean ages of 30.0 and 29.0 years, respectively. Approximately 60% of participants in both groups were female.
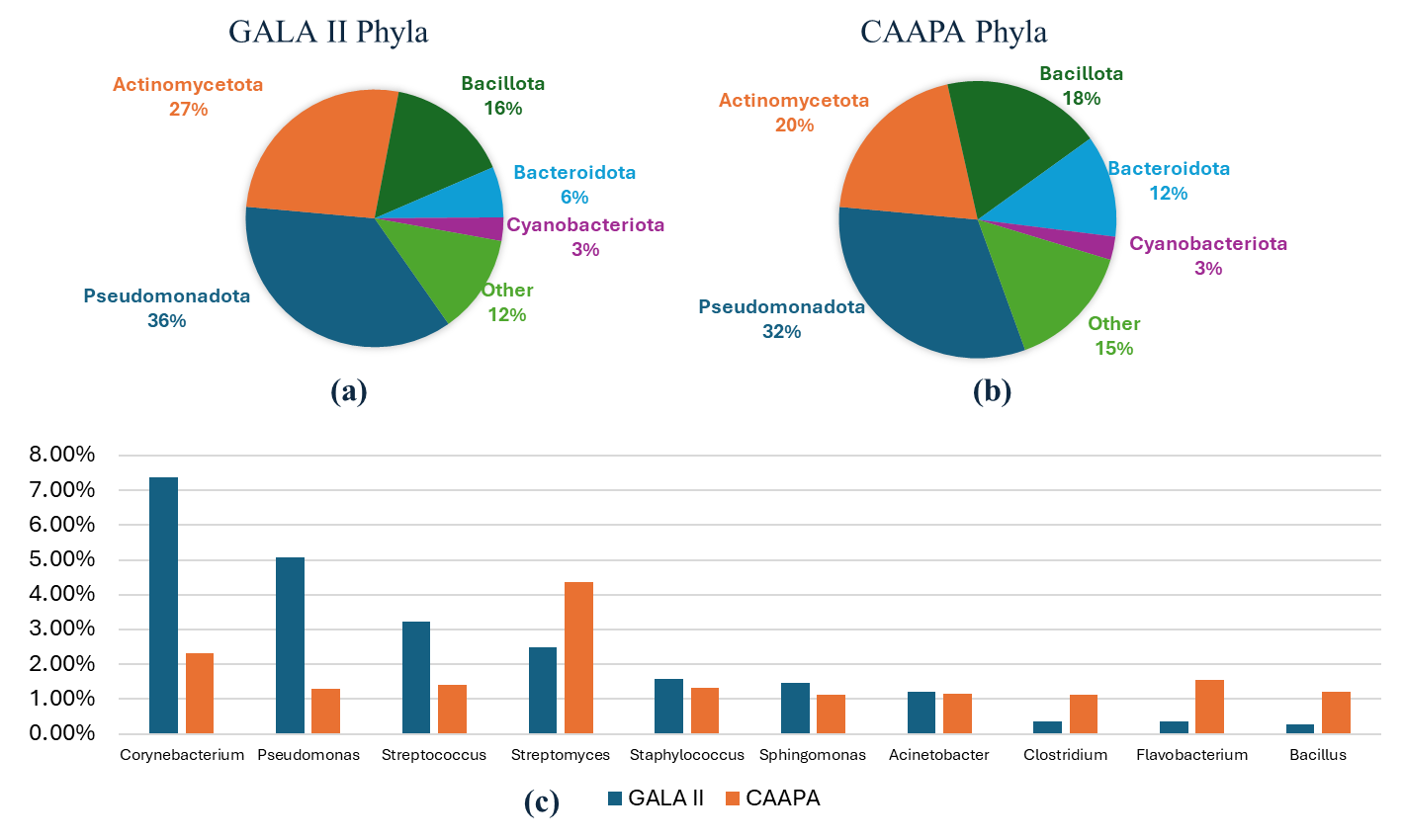
A total of 1084 and 4278 unique species were detected in at least one sample in the GALA II study data and CAAPA study data, respectively. In the GALA II study data, the five species with the highest relative abundance detected were
Mycobacterium canettii (65.49%),
Klebsiella pneumoniae (8.88%),
Streptomyces lividans (4.30%),
Xanthomonas euvesicatoria (3.69%), and
Enterococcus faecium (2.79%); in the CAAPA study data,
Mycobacterium canettii (38.26%),
Corynebacterium segmentosum (11.28%),
Corynebacterium glutamicum (6.69%),
Corynebacterium propinquum (5.96%), and
Klebsiella pneumoniae were (5.56%) were the most prevalent species. Both datasets exhibited a similar composition, being made up predominantly of five phyla—
Pseudomonadota,
Actinomycetota,
Bacillota,
Bacteroidota, and
Cyanobacteriota. These five phyla collectively accounted for over 85% of the total relative abundance in both datasets. Moreover, the relative abundances of these five phyla were closely comparable across the two datasets. In both datasets, 7 genera:
Corynebacterium,
Pseudomonas,
Streptococcus,
Streptomyces,
Staphylococcus,
Sphingomonas, and
Acinetobacter had a relative abundance greater than 1%, with each genus ranking among the top 10 most abundant genera ().
. Comparison of Phyla and Genera Composition Between Study Datasets. (<b>a</b>) GALA II’s five most abundant phyla and all other individual phyla < 2%. (<b>b</b>) CAAPA’s five most abundant phyla and every other individual phylum < 2%. (<b>c</b>) Comparison of 10 most abundant genera between GALA II and CAAPA study datasets reveals a similar composition between datasets on a genus level.
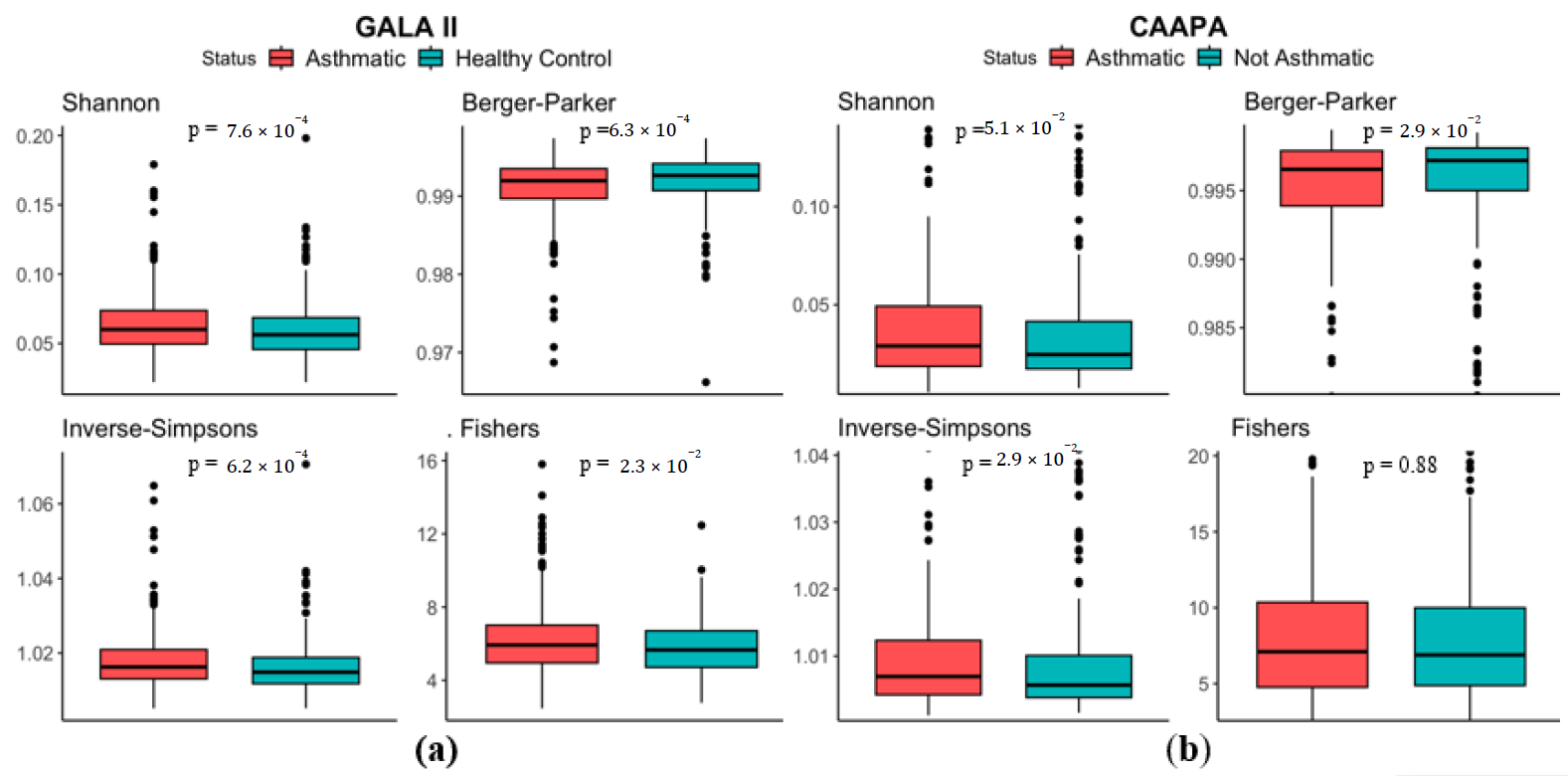
Analysis based on the 694 Hispanic children of the GALA II study showed significant higher Shannon Index (median in asthmatics (
MdnA) = 0.0600, median in healthy controls (
MdnHC) = 0.0562,
p < 0.001), Inverse Simpsons Index (
MdnA = 1.02,
MdnHC = 1.01,
p < 0.001), Fishers index (
MdnA = 5.92,
MdnHC = 5.66,
p = 2.30 × 10
−2), and lower Berger Parker Index (
MdnA = 0.992,
MdnHC = 0.993,
p < 0.001) in samples from the asthmatic group, indicating an increased microbial α diversity in the nasal cavity of asthmatics (a). Results from the 562 CAAPA Participants of African descent further supports this finding, with Berger Parker Index (
MdnA = 0.996,
MdnHC = 0.997,
p = 2.90 × 10
−2) and Inverse Simpsons Index (
MdnA = 1.007,
MdnHC = 1.006,
p = 2.90 × 10
−2) reproducing significant differences, although Shannon Index (
MdnA = 0.0290,
MdnHC = 0.0246,
p = 5.10 × 10
−2) and Fisher’s Index (
MdnA = 7.10,
MdnHC = 6.89,
p = 0.880) presented no statistical significance (b).
. α Diversity comparison between asthmatic and non-asthmatic individuals using Shannon, Berger-Parker, Inverse-Simpsons, and Fishers Indices. (<b>a</b>) In the GALA II study dataset, all four indices indicate greater α diversity among asthmatics and significant differences based on Wilcoxon test. (<b>b</b>) In the CAAPA study dataset, all four indices indicate greater α diversity among asthmatics, but only significant differences in the Berger Parker and Inverse-Simpsons indices.
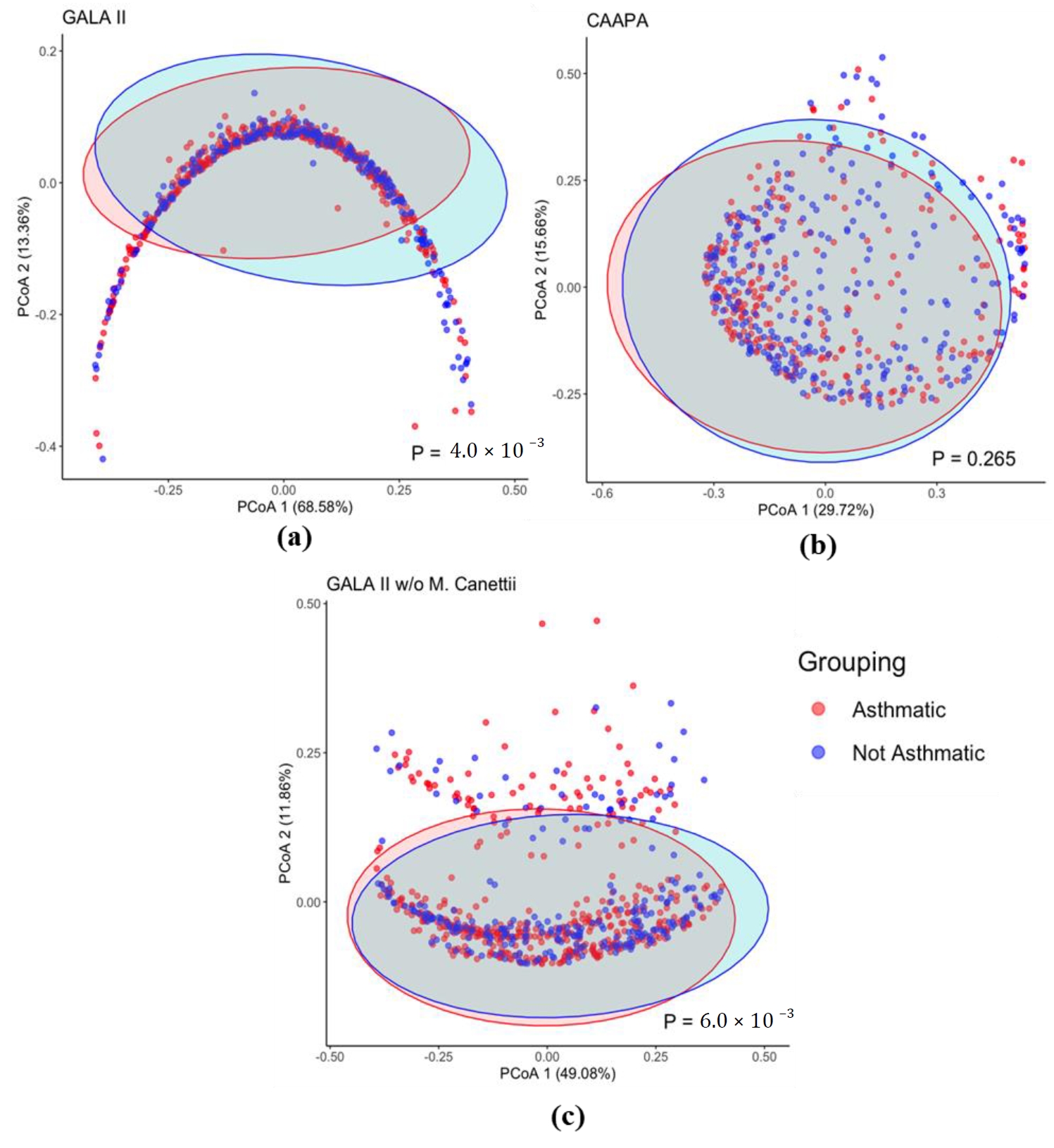
Based on the Bray-Curtis dissimilarity matrix, we also found a significant difference in overall microbiome composition between asthmatics and healthy control patients from GALA II study data (
p = 4.00 × 10
−3), which is not observed, however, in nasal samples from the CAAPA study (
p = 0.265) ().
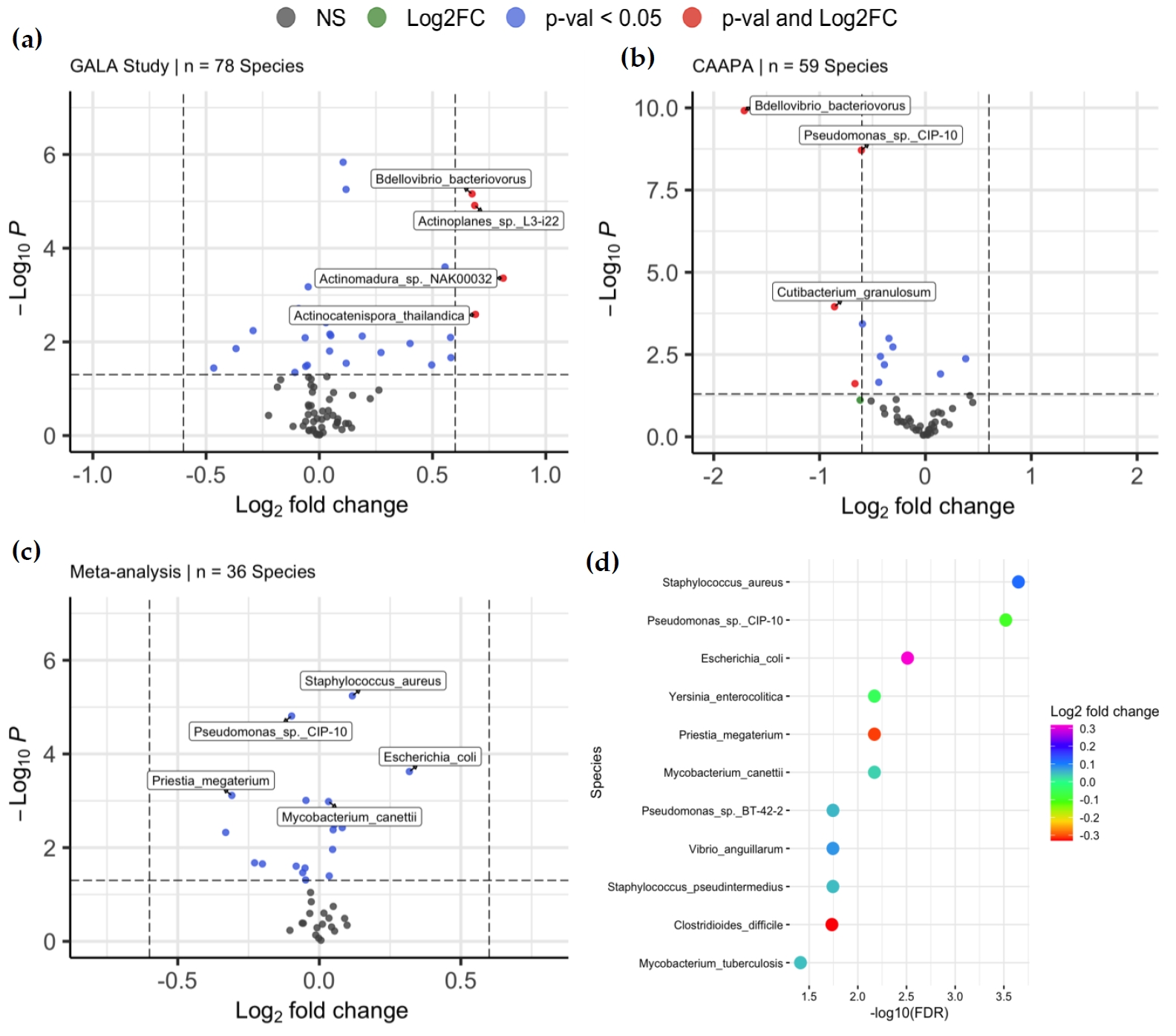
With species of low abundance excluded (mean abundance < 2 and median abundance < 2, Materials and Methods), 78 and 59 species were included in DAA for the GALA II study data and CAAPA study data, respectively. For the GALA II study data, a total of 20 species, such as
Chlorobaculum parvum (log
2FC = 0.105,
Padj = 1.14 × 10
−4),
Staphylococcus pseudintermedius (log
2FC = 0.0466,
Padj = 3.35 × 10
−2),
Staphylococcus aureus (log
2FC = 0.118,
Padj = 1.80 × 10
−4),
Bdellovibrio bacteriovorus (log
2FC = 0.675,
Padj = 1.80 × 10
−4), and
Actinomadura sp.
NAK00032 (log
2FC = 0.812,
Padj = 4.86 × 10
−3) showed a significant difference in abundance between samples from asthmatic and healthy control individuals, with five species negatively enriched and 15 species positively enriched in the asthmatic group compared to the healthy controls. (a and Table S1). Of note, 16 of the 20 species associated with asthma are classified under 2 phyla:
Actinomycetota or
Psudomonadota. For the CAAPA study data, 9 species showed significant differential abundance, and only one species was positively enriched in the asthmatic group compared to healthy control, including
Bdellovibrio bacteriovorus (log
2FC = −1.71,
Padj = 5.87 × 10
−9),
Pseudomonas sp.
CIP-10 (log
2FC = −0.605,
Padj = 4.66 × 10
−8),
Cutibacterium granulosum (log
2FC = −0.859,
Padj = 1.78 × 10
−3),
Clostridium botulinum (log
2FC = −0.345,
Padj = 9.80 × 10
−3), and
Priestia megaterium (log
2FC = −0.595,
Padj = 4.46 × 10
−3) (b and Table S2).
. β Diversity Comparison Between Asthmatic and Non-asthmatic Individuals. (<b>a</b>) The PCoA plot of the GALA II Bray Curtis dissimilarity matrix shows a significant difference between the composition of groups. (<b>b</b>) PCoA plot using CAAPA Bray Curtis dissimilarity matrix reveals no significant difference in composition between groups. (<b>c</b>) Recalculated PCoA plot of the GALA II study dataset after removing the most abundant species <i>Mycobacterium Canettii</i> (65%); the horseshoe shape is no longer present and maintains a significant difference in microbial composition between groups.
. Differentially Abundant Taxa between Asthmatics and Non-asthmatics. (<b>a</b>) Volcano plots of 78 species were included in the DAA of the GALA II study dataset, of which 20 were associated with asthma risk. (<b>b</b>) Volcano plots of 59 species were included in the DAA of the CAAPA study dataset, of which 9 species were found to be associated with asthma risk. (<b>c</b>) Volcano plots of 36 species was included in meta-analysis, where 11 species were associated with asthma. (<b>d</b>) The plot shows the relationship between the log<sub>2</sub>-fold change and <i>p</i>-value of each species associated with asthma in the meta-analysis.
Meta-analysis of the two studies was conducted on the 36 species that were common to the differential abundance analysis models of both studies. 11 species, such as
Staphylococcus aureus (log
2FC = 0.117,
Padj = 2.24 × 10
−4),
Mycobacterium canettii (log
2FC = 0.0326,
Padj = 6.75 × 10
−3), and
Escherichia coli (log
2FC = 0.318,
Padj = 3.09 × 10
−3), showed significant differential abundance between asthmatic and healthy control groups, among which 7 were positively enriched in asthmatic samples. Of note,
Pseudomonas sp.
CIP-10, which presented as DAT in both individual analyses, also showed significance in meta-analyses, indicating its robustness across datasets. Interestingly,
Mycobacterium tuberculosis (log
2FC = 0.0470,
Padj = 3.88 × 10
−2) and
Clostridioides_difficile (log
2FC = −0.331,
Padj = 1.84 × 10
−2) were species significant only in the meta-analysis, while 16 species with significance in either GALA II study data or CAAPA study data showed no statistical significance in meta-analysis, including
Chlorobaculum parvum,
Actinoplanes sp.
L3-i22,
Bdellovibrio bacteriovorus,
Streptomyces lydicus,
Cutibacterium_acnes, and
Actinomadura_sp._
NAK00032 (c,d, and Table S3).
4. Discussion
In our study, we conducted a diversity analysis and differential abundance meta-analysis of two independent datasets, GALA II and CAAPA, which revealed a greater intrasample diversity within asthmatics compared to non-asthmatic patients and identified 22 species significantly associated with asthma. However, there was no indication of a difference in intersample diversity between groups.
The true relationship between the nasal microbiome and asthma is still unclear, partly due to the limited sample size of previous studies. However, our results are consistent with multiple previous studies that reported higher α nasal microbial diversity in asthma patients. For example, Zhou et al. examined in 102 samples the association of nasal microbial diversity with the yellow zone (a period of early loss of the control of asthma). They found a significantly higher Shannon’s Index in yellow zone samples [
16]. In another study of nasal brushings from 72 participants, α diversity measured by Faith’s phylogenic diversity index, which focuses on the richness of evolutionary lineages, indicated a positive, but not statistically significant, trend between nasal bacterial α diversity and asthma activity [
32]. With a much larger sample size, our results extended these findings to four other indices, including Shannon, Berger-Parker, Simpsons, and Fishers, which are more robust and reliable.
Interestingly, even though the PERMANOVA test (
p = 4.00 × 10
−3) for β diversity in the GALA II study data suggested a significant difference between groups, the PcoA plot presented a horseshoe-like shape lacking distinct clustering of groups. This indicates the presence of the horseshoe effect, a common limitation potentially resulting from the extreme variability in the abundance data. It is most often observed in dimension-reducing strategies, resulting from an inability to discriminate among samples that don’t share common features [
33]. In a distance matrix, while Euclidean distance typically increases linearly with the gradient, it often becomes saturated once it surpasses a certain threshold. At extreme values, even when substantial dissimilarity exists between samples, the Euclidean distance metric may represent them as equally dissimilar [
33]. As we recalculated the distance matrix with the top species
Mycobacterium canettii excluded, which had originally accounted for on average a disproportionate 64% of the total species abundance within a sample, the PcoA plot no longer shows indication of the horseshoe effect, while PERMANOVA still revealed a significantly different β diversity between groups (
p = 6.00 × 10
−3) (c).
Equally important, we also revealed taxa associated with asthma at the species level, among which several have never been reported before. For example,
Pseudomonas sp.
CIP-10 showed significant enrichment in both individual DAAs and the combined meta-analysis DAA. To date, little is known about its role in physiology and disease. On the other hand,
Pseudomonas,
p. aeruginosa, another species in its genus level, has been well documented for its role in chronic lung infections, including asthma [
34], where it may induce inflammation and contribute to microbial dysbiosis in the lungs [
35]. Our meta-analysis also identified
Mycobacterium canettii and
Mycobacterium tuberculosis as species associated with asthma. Both species belong to the
Mycobacterium tuberculosis Complex, a group of closely related bacteria that can act as opportunistic pathogens for tuberculosis in humans and animals [
36,
37]. Other members of this complex include
M. bovis,
M. microti,
M. africanum,
M. pinnipedii, and
M. caprae. Other “nontuberculous”
Mycobacterium species (NTM) are increasingly recognized for their role in human disease, such as
M. leprae, which is known to cause leprosy in humans. These NTMs have also been linked to asthma. A study indicates that NTM infections occur nearly 100 times more frequently in asthmatic populations (1.7%) compared to the general population (0.017%). Given the significant role
Mycobacterium tuberculosis complex species already play in the development of diseases like tuberculosis, as well as the association between NTM species and asthma development,
M. tuberculosis and
M. canettii may exhibit similar pathogenic behavior in relation to asthma. However, further research is needed to confirm this potential linkage and elucidate the underlying mechanisms [
38].
In addition, some other notable species from the
Staphylococcus genus, specifically
S. aureus,
S. homonis and
S. pseudintermedius, are known opportunistic pathogens of asthma [
39,
40,
41]. For example,
S. aureus infections often induce or intensify inflammatory responses, contributing significantly to the development of allergic asthma [
42]. Notably, species from the
Moraxella and
Haemophilus genera, though previously linked to asthma, were not found to be associated with asthma risk in our study [
43,
44,
45,
46,
47]. Future research could benefit from additional analyses, such as functional pathway analysis, to better understand the mechanisms underlying the role of these species. In summary, our research highlights a greater microbial diversity among asthmatic patients. It identifies established and emerging microbial species linked to asthma, underscoring the need for further investigation and refinement in this field.
However, some limitations still exist within our study. Firstly, while α and β diversity measures can be used as possible indicators for early asthma detection by measuring microbiota dysbiosis during infancy or as a guide for tailoring antibiotic treatments aimed at reducing microbial diversity, the clinical significance of our results remains limited [
47]. This is because of the small differences in median alpha diversity between groups, making a clear interpretation in clinical settings challenging to distinguish, as well as the lack of robustness across beta diversity findings. Second, our analysis focused exclusively on the presence of asthma. Given that previous research has highlighted the importance and greater heterogeneity of asthma endotypes, future studies should explore the associations between the nasal microbiome and different asthma endotypes [
48]. Additionally, while we obtained results consistent with previous studies from two relatively large datasets, both were case-control studies, which may offer limited insights into the relationship between the nasal microbiome and asthma. A prospective study design would help clarify the causal links between specific microbial species and asthma development. Additionally, relying solely on meta-transcriptomics data leaves the mechanisms behind these associations unclear; integrating tools like metabolic analysis could help uncover the potential functional basis.
5. Conclusions
In summary, by employing datasets of larger sample sizes and meta-transcriptomic analytical strategies, our study provides greater insight into the role of the nasal microbiome in asthma risk. Diversity analysis reveals a greater α diversity within asthmatic patients as compared to healthy control patients but no difference in β diversity between groups. Meta-analysis of two individual differential abundance analysis identifies 20 species associated with asthma risk. Our findings will hopefully facilitate further meta-transcriptomic research in the nasal microbiome, specifically exploring the species associated with asthma that have not been widely studied or reported.
Supplementary Materials
The following supporting information can be found at: https://www.sciepublish.com/article/pii/328, Table S1: Differentially abundant taxa (DATs) in GALA II study dataset. Table S2: Differentially abundant taxa (DATs) in CAAPA study dataset. Table S3: Differentially abundant taxa (DATs) in meta-analysis of GALA II and CAAPA study datasets.
Author Contributions
A.L., M.Y. and X.Y. conducted the research. The study conception and design were contributed by M.Y. and W.C., M.Y., X.Y. and W.C. provided supervision for the research. Clinical expertise was provided by X.Y., K.G. and A.F.W.-E. The initial draft of the manuscript was prepared by A.L. and M.Y., and all authors participated in critical revision of the manuscript for significant intellectual content. All authors have reviewed and approved the final version of the manuscript for submission.
Ethics Statement
Not applicable.
Informed Consent Statement
Not applicable.
Funding
A.W-E was supported in part by a NCATS NIH grant (KL2TR001856).
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
1.
Campbell CD, Gleeson M, Sulaiman I. The role of the respiratory microbiome in asthma.
Front. Allergy 2023,
4, 1120999.
[Google Scholar]
2.
Shin YH, Hwang J, Kwon R, Lee SW, Kim MS, Jajarmi M, et al. Global, regional, and national burden of allergic disorders and their risk factors in 204 countries and territories, from 1990 to 2019: A systematic analysis for the Global Burden of Disease Study 2019.
Allergy 2023,
78, 2232–2254.
[Google Scholar]
3.
Zhou X, Sampath V, Nadeau KC. Effect of air pollution on asthma.
Ann. Allergy Asthma Immunol. 2024,
132, 426–432.
[Google Scholar]
4.
Georas SN, Khurana S. Update on asthma biology.
J. Allergy Clin. Immunol. 2024,
153, 1215–1228.
[Google Scholar]
5.
Ntontsi P, Photiades A, Zervas E, Xanthou G, Samitas K. Genetics and Epigenetics in Asthma.
Int. J. Mol. Sci. 2021,
22, 2412.
[Google Scholar]
6.
Varricchi G, Brightling CE, Grainge C, Lambrecht BN, Chanez P. Airway remodelling in asthma and the epithelium: On the edge of a new era.
Eur. Respir. J. 2024,
63, 2301619.
[Google Scholar]
7.
Aggarwal N, Kitano S, Puah GRY, Kittelmann S, Hwang IY, Chang MW. Microbiome and Human Health: Current Understanding, Engineering, and Enabling Technologies.
Chem. Rev. 2023,
123, 31–72.
[Google Scholar]
8.
Van Every H, Franzosa EA, Nguyen LH, Huttenhower C. Microbiome epidemiology and association studies in human health.
Nat. Rev. Genet. 2023,
24, 109–124.
[Google Scholar]
9.
Man WH, de Steenhuijsen Piters WAA, Bogaert D. The microbiota of the respiratory tract: Gatekeeper to respiratory health.
Nat. Rev. Microbiol. 2017,
15, 259–270.
[Google Scholar]
10.
Sokolowska M, Frei R, Lunjani N, Akdis CA, O’Mahony L. Microbiome and asthma.
Asthma Res. Pract. 2018,
4, 1.
[Google Scholar]
11.
Barcik W, Boutin RCT, Sokolowska M, Finlay BB. The Role of Lung and Gut Microbiota in the Pathology of Asthma.
Immunity 2020,
52, 241–255.
[Google Scholar]
12.
Biesbroek G, Tsivtsivadze E, Sanders EAM, Montijn R, Veenhoven RH, Keijser BJF, et al. Early respiratory microbiota composition determines bacterial succession patterns and respiratory health in children.
Am. J. Respir. Crit. Care Med. 2014,
190, 1283–1292.
[Google Scholar]
13.
Teo SM, Mok D, Pham K, Kusel M, Serralha M, Troy N, et al. The Infant Nasopharyngeal Microbiome Impacts Severity of Lower Respiratory Infection and Risk of Asthma Development.
Cell Host Microbe Rev. 2015,
17, 704–715.
[Google Scholar]
14.
McCauley KE, Flynn K, Calatroni A, DiMassa V, LaMere B, Fadrosh DW, et al. Seasonal airway microbiome and transcriptome interactions promote childhood asthma exacerbations.
J. Allergy Clin. Immunol. 2022,
150, 204–213.
[Google Scholar]
15.
Depner M, Ege MJ, Cox MJ, Dwyer S, Walker AW, Birzele LT, et al. Bacterial microbiota of the upper respiratory tract and childhood asthma.
J. Allergy Clin. Immunol. 2017,
139, 826–834.e13.
[Google Scholar]
16.
Zhou Y, Jackson D, Bacharier LB, Mauger D, Boushey H, Castro M, et al. The upper-airway microbiota and loss of asthma control among asthmatic children.
Nat. Commun. 2019,
10, 5714.
[Google Scholar]
17.
Avalos-Fernandez M, Alin T, Métayer C, Thiébaut R, Enaud R, Delhaes L. The respiratory microbiota alpha-diversity in chronic lung diseases: First systematic review and meta-analysis.
Respir. Res. 2022,
23, 214.
[Google Scholar]
18.
Tang HHF, Lang A, Teo SM, Judd LM, Gangnon R, Evans MD, et al. Developmental patterns in the nasopharyngeal microbiome during infancy are associated with asthma risk.
J. Allergy Clin. Immunol. 2021,
147, 1683–1691.
[Google Scholar]
19.
Genes-environments and Admixture in Latino Asthmatics (GALA II) GWAS Study. Available online: https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs001274.v2.p1 (accessed on 30 September 2024).
20.
Consortium on Asthma among African-ancestry Populations in the Americas (CAAPA). Available online: https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs001123.v2.p1 (accessed on 30 September 2024).
21.
Ewels PA, Peltzer A, Fillinger S, Patel H, Alneberg J, Wilm A, et al. The nf-core framework for community-curated bioinformatics pipelines.
Nat. Biotechnol. 2020,
38, 276–278.
[Google Scholar]
22.
Stamouli S, Beber ME, Normark T, Christensen TA, Andersson-Li L, Borry M, et al. nf-core/taxprofiler: Highly parallelised and flexible pipeline for metagenomic taxonomic classification and profiling. bioRxiv 2023, submitted.
23.
Chen S, Zhou Y, Chen Y, Gu J. fastp: An ultra-fast all-in-one FASTQ preprocessor.
Bioinformatics 2018,
34, i884–i890.
[Google Scholar]
24.
NCBI. Homo Sapiens Genome Assembly GRCh38. Available online: https://www.ncbi.nlm.nih.gov/data-hub/assembly/GCF_000001405.26/ (accessed on 1 October 2024).
25.
Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome.
Genome. Biol. 2009,
10, R25.
[Google Scholar]
26.
Wood DE, Lu J, Langmead B. Improved metagenomic analysis with Kraken 2.
Genome. Biol. 2019,
20, 257.
[Google Scholar]
27.
Loman Lab Mock Community Experiments. Available online: https://lomanlab.github.io/mockcommunity/mc_databases.html (accessed on 30 September 2024).
28.
Walters KE, Martiny JBH. Alpha-, beta-, and gamma-diversity of bacteria varies across habitats.
PLoS ONE 2020,
15, e0233872.
[Google Scholar]
29.
Oksanen J, Simpson GL, Blanchet FG, Kindt R, Legendre P, Minchin PR, et al. vegan: Community Ecology Package. Available online: https://cran.r-project.org/web/packages/vegan/index.html (accessed on 30 September 2024).
30.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2.
Genome. Biol. 2014,
15, 550.
[Google Scholar]
31.
Viechtbauer W. Conducting Meta-Analyses in R with the metafor Package.
J. Stat. Softw. 2010,
36, 1–48.
[Google Scholar]
32.
Fazlollahi M, Lee TD, Andrade J, Oguntuyo K, Chun Y, Grishina G, et al. The Nasal Microbiome in Asthma.
J. Allergy Clin. Immunol. 2018,
142, 834–843.e2.
[Google Scholar]
33.
Morton JT, Toran L, Edlund A, Metcalf JL, Lauber C, Knight R. Uncovering the Horseshoe Effect in Microbial Analyses.
mSystems 2017,
2, e00166-e16.
[Google Scholar]
34.
Zhang Q, Illing R, Hui CK, Downey K, Carr D, Stearn M, et al. Bacteria in sputum of stable severe asthma and increased airway wall thickness.
Respir. Res. 2012,
13, 35.
[Google Scholar]
35.
Garcia-Clemente M, de la Rosa D, Máiz L, Girón R, Blanco M, Olveira C, et al. Impact of Pseudomonas aeruginosa Infection on Patients with Chronic Inflammatory Airway Diseases.
J. Clin. Med. 2020,
9, 3800.
[Google Scholar]
36.
Mansfield KG, Fox JG. Chapter 16-Bacterial Diseases. In The Common Marmoset in Captivity and Biomedical Research, 1st ed.; Marini R, Wachtman L, Tardif S, Mansfield K, Fox J, Eds.; Academic Press: Cambridge, MA, USA, 2019; pp. 265–287.
37.
Blouin Y, Cazajous G, Dehan C, Soler C, Vong R, Hassan MO, et al. Progenitor “Mycobacterium canettii” Clone Responsible for Lymph Node Tuberculosis Epidemic, Djibouti.
Emerg. Infect. Dis. 2014,
20, 21–28.
[Google Scholar]
38.
Fritscher LG, Marras TK, Bradi AC, Fritscher CC, Balter MS, Chapman KR. Nontuberculous Mycobacterial Infection as a Cause of Difficult-to-Control Asthma: A Case-Control Study.
Chest 2011,
139, 23–27.
[Google Scholar]
39.
Small C, Beatty N, El Helou G. Staphylococcus pseudintermedius Bacteremia in a Lung Transplant Recipient Exposed to Domestic Pets.
Cureus 2021,
13, e14895.
[Google Scholar]
40.
Hilty M, Burke C, Pedro H, Cardenas P, Bush A, Bossley C, et al. Disordered Microbial Communities in Asthmatic Airways.
PLoS ONE 2010,
5, e8578.
[Google Scholar]
41.
Frank DN, Feazel LM, Bessesen MT, Price CS, Janoff EN, Pace NR. The human nasal microbiota and Staphylococcus aureus carriage.
PLoS ONE 2010,
5, e10598.
[Google Scholar]
42.
Jorde I, Schreiber J, Stegemann-Koniszewski S. The Role of Staphylococcus aureus and Its Toxins in the Pathogenesis of Allergic Asthma.
Int. J. Mol. Sci. 2022,
24, 654.
[Google Scholar]
43.
Bogaert D, Keijser B, Huse S, Rossen J, Veenhoven R, van Gils E, et al. Variability and Diversity of Nasopharyngeal Microbiota in Children: A Metagenomic Analysis.
PLoS ONE 2011,
6, e17035.
[Google Scholar]
44.
Bar K, Litera-Bar M, Sozańska B. Bacterial Microbiota of Asthmatic Children and Preschool Wheezers’ Airways—What Do We Know?
Microorganisms 2023,
11, 1154.
[Google Scholar]
45.
Chung KF. Airway microbial dysbiosis in asthmatic patients: A target for prevention and treatment?
J. Allergy Clin. Immunol. 2017,
139, 1071–1081.
[Google Scholar]
46.
Zhou Y, Gao H, Mihindukulasuriya KA, La Rosa PS, Wylie KM, Vishnivetskaya T, et al. Biogeography of the ecosystems of the healthy human body.
Genome. Biol. 2013,
14, R1.
[Google Scholar]
47.
Valverde-Molina J, García-Marcos L. Microbiome and Asthma: Microbial Dysbiosis and the Origins, Phenotypes, Persistence, and Severity of Asthma.
Nutrients 2023,
15, 486.
[Google Scholar]
48.
Gans MD, Gavrilova T. Understanding the immunology of asthma: Pathophysiology, biomarkers, and treatments for asthma endotypes.
Paediatr. Respir. Rev. 2020,
36, 118–127.
[Google Scholar]