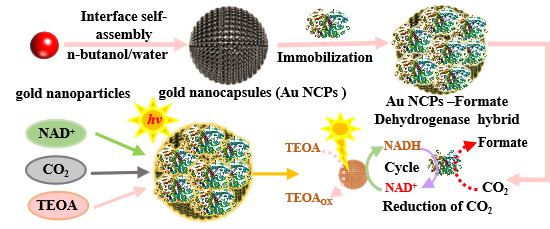
1. Introduction
The excessive reliance on fossil fuels in current human activities has resulted in excessive carbon dioxide (CO
2) emissions, leading to a range of potential ecological, physical, and health impacts [
1,
2]. Therefore, efficient and green methods should be developed to achieve CO
2 recycling in order to address CO
2 pollution and attain the goal of carbon neutrality as soon as possible. In recent years, the use of dehydrogenase cascade catalysis to convert CO
2 into compounds like formate and methanol has been extensively studied [
3,
4]. The enzymatic conversion of CO
2 to formic acid is a crucial step in these cascade reactions [
3,
5]. However, the high cost of dehydrogenases and the stoichiometric consumption of cofactors make practical carbon fixation using dehydrogenases unfeasible [
6]. Therefore, it is necessary to develop efficient enzyme immobilization and cofactor regeneration systems, coupling enzymatic CO
2 reduction with cofactor regeneration for efficient operation [
7].
Mimicking natural photosynthesis and using unlimited renewable solar energy to produce chemical fuels and other synthetic energy from CO
2 is one of the most promising solutions for high-value CO
2 conversion [
8,
9]. This artificial photosynthesis employs photocatalysts to immobilize dehydrogenases, creating photo-enzyme hybrids. These systems can catalyze the conversion of CO
2 into high-value compounds under visible light, using photo-catalytically regenerated NAD(P)H as the “energy currency” for enzymatic catalysis [
10]. This approach integrates the excellent light-absorption properties of photocatalysts with the high activity and selectivity of enzymes [
11].
However, due to the poor photostability and high cost of NADH, to achieve continuous photocatalytic CO
2 reduction through photo-enzyme hybrids, an efficient and straightforward method for NADH recycling is essential [
12,
13]. Although many efficient photocatalytic NADH regeneration methods have been developed, most of these methods rely on organic electron mediators ([Cp
∗Rh(bpy)H
2O]
2+ and [Cp
∗RhCl
2]
2) to transfer electrons [
14,
15]. However, these electron mediators are expensive and can inhibit each other with FDH, significantly reducing enzyme activity [
16,
17]. Therefore, developing NADH regeneration technology that does not rely on exogenous electron mediators is particularly important for the sustainability of photo-enzyme coupling for carbon fixation. Moreover, immobilizing photocatalysts and enzymes on a carrier to construct a photo-enzyme hybrid system can help improve enzyme stability and the potential for reuse [
18]. However, the free radical toxicity of photocatalysts makes this photo-enzyme hybrid system less stable [
19]. Utilizing photocatalysts directly as enzyme carriers can simplify the photo-enzyme hybrid system, reduce the spatial distance between the photocatalysts and enzymes, enhance photoelectron transfer efficiency, and, in turn, promote enzyme-catalyzed reactions. Therefore, the choice and design of photocatalysts (enzyme carriers) are crucial for constructing photo-enzyme hybrid systems. Research indicates that gold nanocages, which are hollow cage-like nanomaterials assembled from gold nanocapsules (Au NCPs) with full-spectrum absorption characteristics, exhibit high photocatalytic efficiency due to their electromagnetic field enhancement effect [
20]. Their high biocompatibility and hollow structure suggest potential for serving as both photocatalysts and enzyme carriers.
In this study, we employed Au NCPs as both the photocatalysts and the carriers for formate dehydrogenase (FDH). Initially, we investigated the protective performance of Au NCPs towards NADH under illumination and optimized their photocatalytic activity for NADH regeneration without external electron mediators. Subsequently, we constructed a light-enzyme hybrid system using the Au NCPs and FDH, optimizing the conditions for CO
2 reduction within this hybrid system. This approach achieved efficient light-driven CO
2 reduction to formate using the Au NCPs-FDH hybrid system, providing a solid foundation for developing photo-enzyme coupling hybrid systems and advancing light-driven enzyme catalysis for efficient carbon fixation.
2. Materials and Methods
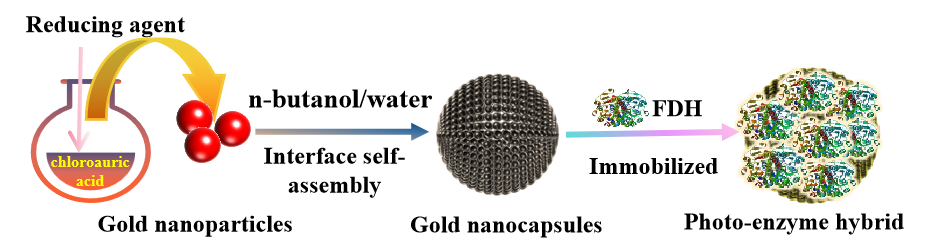
The schematic diagram of the preparation of gold nanoparticles, gold nanocapsules, and the immobilization of formate dehydrogenase (FDH) to construct the photo-enzyme hybrid system is shown in Scheme 1.
Scheme 1. Schematic diagram of the preparation of gold nanocapsules and the immobilization of FDH on the gold nanocapsules.
Gold nanoparticles (Au NPs) preparation: Following the method reported by Li et al. [
21], Au NPs were synthesized via the sodium citrate reduction of chloroauric acid. The pH of the reaction solution was adjusted to 2 using 2 M hydrochloric acid. The Au NPs were then precipitated by adding isopropanol in a 1:1 volume ratio with the reaction solution. The precipitated Au NPs were washed sequentially three times each with methanol and n-butanol. Finally, the Au NPs were resuspended in ultrapure water and concentrated to obtain a 20 wt% suspension.
Synthesis of gold nanocapsules (Au NCPs): Following previous reports [
20], a suspension of Au NPs and n-butanol were mixed in various volume ratios to create two distinct phases with a clear oil-water interface. The aqueous phase was emulsified in n-butanol through vortex shaking for 60 s and ultrasonication for 5 min. The resulting emulsion, encapsulating the gold nano colloids, was left to stand for 12 h, allowing the gold particles to self-assemble into Au NCPs at the oil-water interface. The Au NCPs were then separated by centrifugation, washed sequentially three times with n-butanol and ethanol, freeze-dried for 24 h, and stored at 4 °C for future use.
The morphology and cavity structure of the Au NCPs were observed using a transmission electron microscope (JEOL JEM 2100F, Tokyo, Japan). In contrast, the external structure was observed using a scanning electron microscope (Hitachi SU8600 SEM, Tokyo, Japan). The UV-vis spectra of both Au NPs and Au NCPs were measured using a TECAN microplate reader.
2.2. Photocatalytic Regeneration of NADH by Au NCPs
Protective properties of Au NCPs against NADH under light: NADH is known to be less stable under light and acidic conditions [
22]. The effects of the electron donor triethanolamine (TEOA) and Au NCPs on the stability of NADH under light were first tested. A 2 mL reaction system containing 0.50 mM NADH, Au NCPs (1.25 mg/mL), and TEOA (100 mM) was maintained at a controlled temperature of 30 °C and irradiated with 50 mW/cm
2 LED white light. Samples were taken at 0, 1, 2, 4, 6, and 8 h. The remaining concentration of NADH was calculated by measuring its absorbance at 340 nm [
23] to evaluate the protective performance of the Au NCPs on NADH under light exposure.
Photocatalytic NADH Regeneration: The multichannel photocatalytic reaction system (WATTCAS) was used to conduct the photocatalytic NADH regeneration experiments. The photocatalytic system containing NAD
+ (0.50 mM), TEOA, and Au NCPs (1.25 mg/mL) was exposed to LED white light for a certain period. The supernatant was periodically sampled to measure the concentration of NADH, and the regeneration yield of NADH was calculated using Equation (1).
2.3. Immobilization of FDH on Au NCPs for the Photo-Enzymatic Hybrid System
The FDH (0.10 mg/mL) was mixed with Au NCPs (1.50 mg/mL) in a buffer solution (100 mM, pH = 7.40) and shaken the mixture at a set temperature for a certain period. The protein concentration in the supernatant was measured using the Bradford assay, and the immobilization yield of FDH was calculated (Equation (2)). The photo-enzyme hybrid system was centrifuged at 8000 rpm for 5 minutes, and the precipitate was collected and washed twice with a buffer solution. Then, freeze-dry the precipitate and store it at 4 °C for future use. The activity of FDH was assessed by measuring the initial reduction rates of formate catalyzed by both free FDH and the photo-enzyme hybrid [
9]. The free FDH, sodium formate (1 mM), and NAD
+ (10 mM) were added to PBS and shaken at room temperature for 3 min, and the absorbance at 340 nm was measured to determine free enzyme activity. The immobilized enzyme activity was evaluated by replacing the free FDH with the photo-enzyme hybrid while maintaining all other conditions constant. The recovery of enzyme activity was calculated according to Equation (3).
where
C0 is the initial concentration of FDH (μg/mL);
C is the residual protein concentration in the supernatant after the immobilization reaction (μg/mL).
where
A1 and
A are the absorbance at 340 nm of the immobilized and free FDH, respectively, after their reaction with the substrate.
2.4. Light-Driven Photo-Enzyme Hybrid System for Converting CO2 to Formate
The reaction solution included NAD
+ (0.50 mM), free FDH or Au NCPs—FDH hybrid (0.50 mg/mL) sodium molybdate, and TEOA (300 mM) dispersed into 2 mL of PBS (100 mM, pH = 7.40), and the reaction proceeded at 30 °C. Before the reaction, the system was purged with CO
2 (99.99%) for 5 min, then sealed with 200 mL of CO
2 gas bag, and then the reaction was carried out under the irradiation of LED white light (150 mW/cm
2). The formate concentration in the supernatant was determined by high-performance liquid chromatography (Agilent, Santa Clara, CA, USA) with a UV detector.
3. Results and Discussion
3.1. Morphological Structure and Light Absorption Properties of Gold Nanocapsules
a shows the TEM image of the gold nanocapsules (Au NCPs), which were self-assembled from Au nanoparticles (Au NPs). The Au NCPs are presented as spherical structures, with a small number of Au nanoparticles at their edges, having an average particle size of 13.06 ± 2.49 nm (b,c). The HAADF-STEM image (e) and the SEM image (f) confirm that the internal structure of the obtained Au NCPs is hollow, and a number of boundary and stacking defects can be observed on their surfaces, the existence of which is essentially necessary for the topological constraints on the spherical surface [
20,
24]. Additionally, f shows the UV-Vis absorption spectra of both Au NPs and Au NCPs. The red Au NPs dispersed in water (inset of f) exhibit a characteristic LSPR peak centered at 530 nm. In contrast, the Au NCPs (black dashed line in f) do not display a peak at 530 nm; instead, they show a broad near-infrared plasmon absorption band, which is attributed to the strong plasmonic coupling between the Au NPs in the Au capsule shell [
25]. As a result, the obtained Au NCPs are dark black in color and exhibit enhanced absorption capacity for visible light.
. (<b>a</b>) Transmission electron microscopy (TEM) images of gold nanocapsules (Au NCPs); (<b>b</b>) TEM images of gold nanoparticles (Au NPs) assembled into Au NCPs; (<b>c</b>) Particle size distribution of Au NPs; (<b>d</b>) HAADF-STEM images of Au NCPs; (<b>e</b>) Scanning electron micrographs (SEM) of Au NCPs; (<b>f</b>) UV-vis absorption spectra of Au NCPs and Au NPs, the inset is a physical image of Au NPs and Au NCPs.
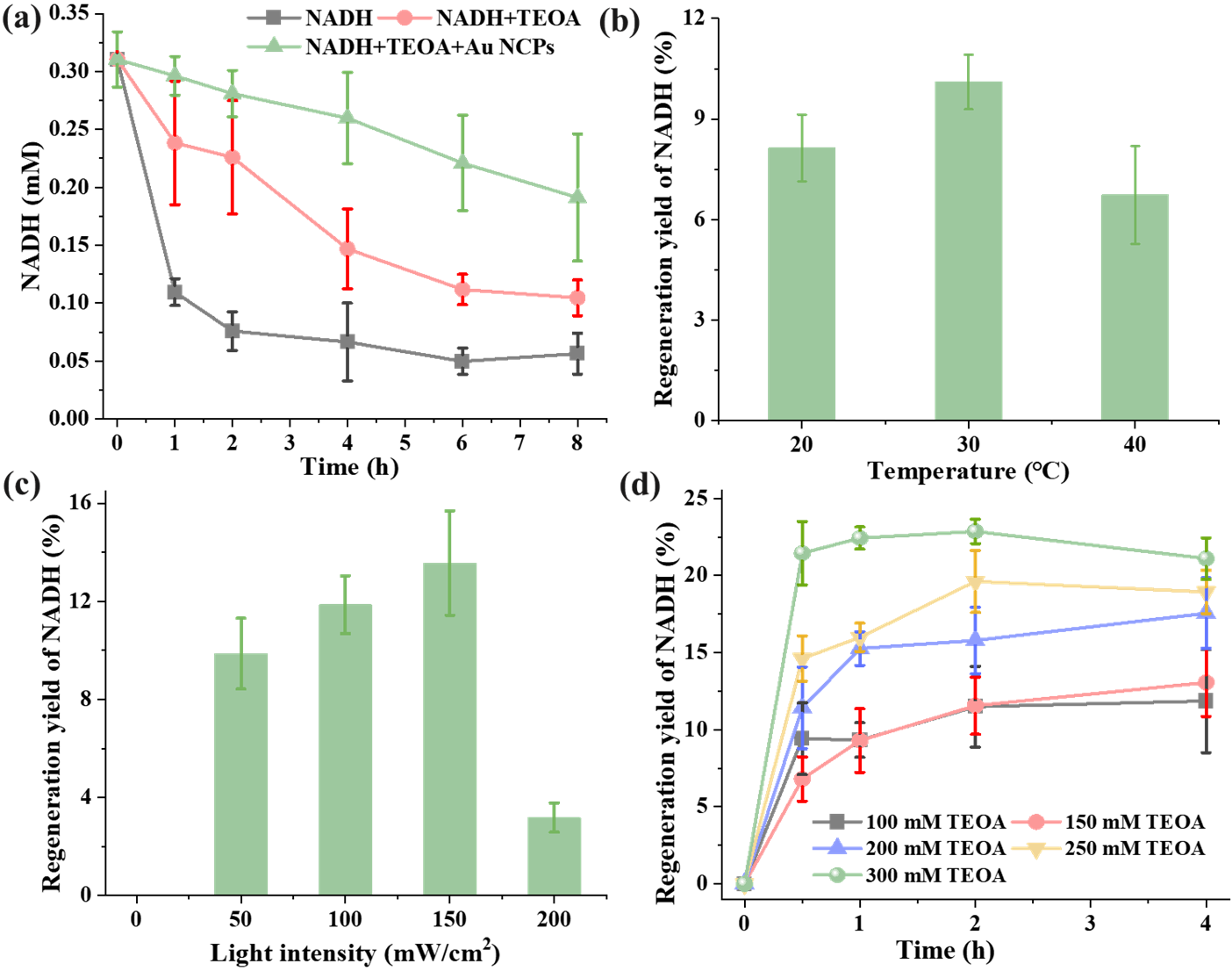
The reduced coenzyme NADH serves as a critical cofactor for formate dehydrogenase (FDH) in catalyzing the CO
2 reduction reaction [
12,
26]. Previous studies have demonstrated that NADH is highly unstable and readily loses its biological activity when exposed to light [
22]. Consequently, we first examined the protective properties of the electron donor triethanolamine (TEOA) and Au NCPs against NADH degradation under light exposure. As illustrated in a, the concentration of NADH exhibited a marked decline of 78.6% following exposure to 50 mW/cm
2 white light for a period of four hours. In contrast, the concentration of NADH decreased by only 52.70% upon the addition of TEOA and illumination for the same duration. The increased stability of NADH in the TEOA system can be attributed to the alkaline nature of the TEOA and the higher stability of NADH molecules in alkaline environments [
22,
27]. Moreover, the concentration of NADH in the light system containing both TEOA and Au NCPs decreased by only 16.33% under identical conditions. This result indicates that NADH exhibits greater stability in the light system with Au NCPs. This enhanced stability is likely due to the excellent full-spectrum absorption properties of the Au NCPs, which effectively shield some of the photons from exerting detrimental effects on NADH molecules. Therefore, it can be concluded that the combined effect of TEOA and Au NCPs prolongs the stabilization of NADH under light illumination.
Then, using TEOA as an electron donor, we examined the performance of temperature on the photocatalytic regeneration of NADH by Au NCPs. As shown in b, the photocatalytic regeneration yield of NADH by Au NCPs initially increased and then decreased with rising temperature. The highest regeneration yield of NADH was observed at a reaction temperature of 30 °C, where the NADH regeneration rate reached 10.10%. Since we did not add exogenous electron mediators, such as [Cp
∗Rh(bpy)H
2O]
2+, which is widely used in photocatalytic coenzyme regeneration systems, the Au NCPs regenerated NADH by directly transferring photoelectrons, which resulted in a low regeneration yield of Au NCPs photocatalytic NADH, compared with other photocatalytic systems that relied on the electron mediators to transfer of photoelectrons (regeneration yield = 40~100%) [
23,
28,
29]. This is because the yield of direct photoelectron transfer to regenerate NADH in photocatalytic systems without exogenous electron mediators (regeneration yield = 18.09~22.5%) was generally low according to previous studies [
22,
28]. However, considering the toxicity of organic electron donors to formate dehydrogenase [
16,
17], this direct electron transfer photoreaction system for regenerating coenzymes is more compatible with the FDH enzyme-catalyzed reaction.
We further optimized the light intensity and electron donor concentration for the NADH regeneration reaction. As shown in c, under light-avoidance conditions (light intensity of 0 mW/cm²), the Au NCPs showed no regenerative activity toward NADH. The yield of NADH regeneration through Au NCPs photocatalysis initially increased and then decreased as the light intensity increased. The highest NADH regeneration yield was achieved at a light intensity of 150 mW/cm², reaching 13.56%. In addition, with the increase of TEOA concentration from 100 mM to 300 mM, the reaction equilibrium time of Au NCPs photocatalytic regeneration of NADH was shortened from 2 h to 0.5 h, and the regeneration yield of NADH was increased to 22.65% (d). Therefore, the optimal reaction conditions for the photocatalytic regeneration of NADH by Au NCPs were temperature 30 °C, light intensity 150 mW/cm
2, and TEOA concentration 300 mM.
. (<b>a</b>) Concentration change curve of NADH under white light illumination; (<b>b</b>) Regeneration yield of photocatalytic NADH by Au NCPs at different temperatures (Au NCPs: 1.25 mg/mL, TEOA: 100 mM, light intensity: 50 mW/cm<sup>2</sup>, reaction time: four hours); (<b>c</b>) Regeneration yield of photocatalytic NADH by Au NCPs under different light intensities (Au NCPs: 1.25 mg/mL, TEOA: 100 mM, 30 °C, reaction time: four hours); (<b>d</b>) Photocatalytic NADH regeneration yield of Au NCPs at different electron donor (TEOA) concentrations (Au NCPs: 1.25 mg/mL, light intensity: 150 mW/cm<sup>2</sup>, 30 °C).
Furthermore, a scanning electron microscope (SEM) was used to characterize the morphology of the formate dehydrogenase (FDH) hybrids immobilized on Au NCPs. The morphology of the Au NCPs of immobilized FDH did not undergo any notable alterations (a). Energy-dispersive X-ray (EDX) mapping scans revealed a uniform distribution of gold (Au) and molybdenum (Mo) elements in the hybrids (b,c), with the relative mass ratio of Au at 66.34 wt% and Mo at 1.26 wt% (d). Given that the FDH molecule contains a Mo ion active center, as previously reported in references [
30,
31], the uniform distribution of Mo provides strong evidence that the Au NCPs have successfully loaded FDH.
. (<b>a</b>) SEM image of the Au NCPs hybrid with formate dehydrogenase (FDH) immobilized; the EDX mapping of gold (<b>b</b>) and molybdenum (<b>c</b>) in the Au NCPs hybrid with FDH immobilized; (<b>d</b>) the relative mass content of various elements on the surface of the hybrids.
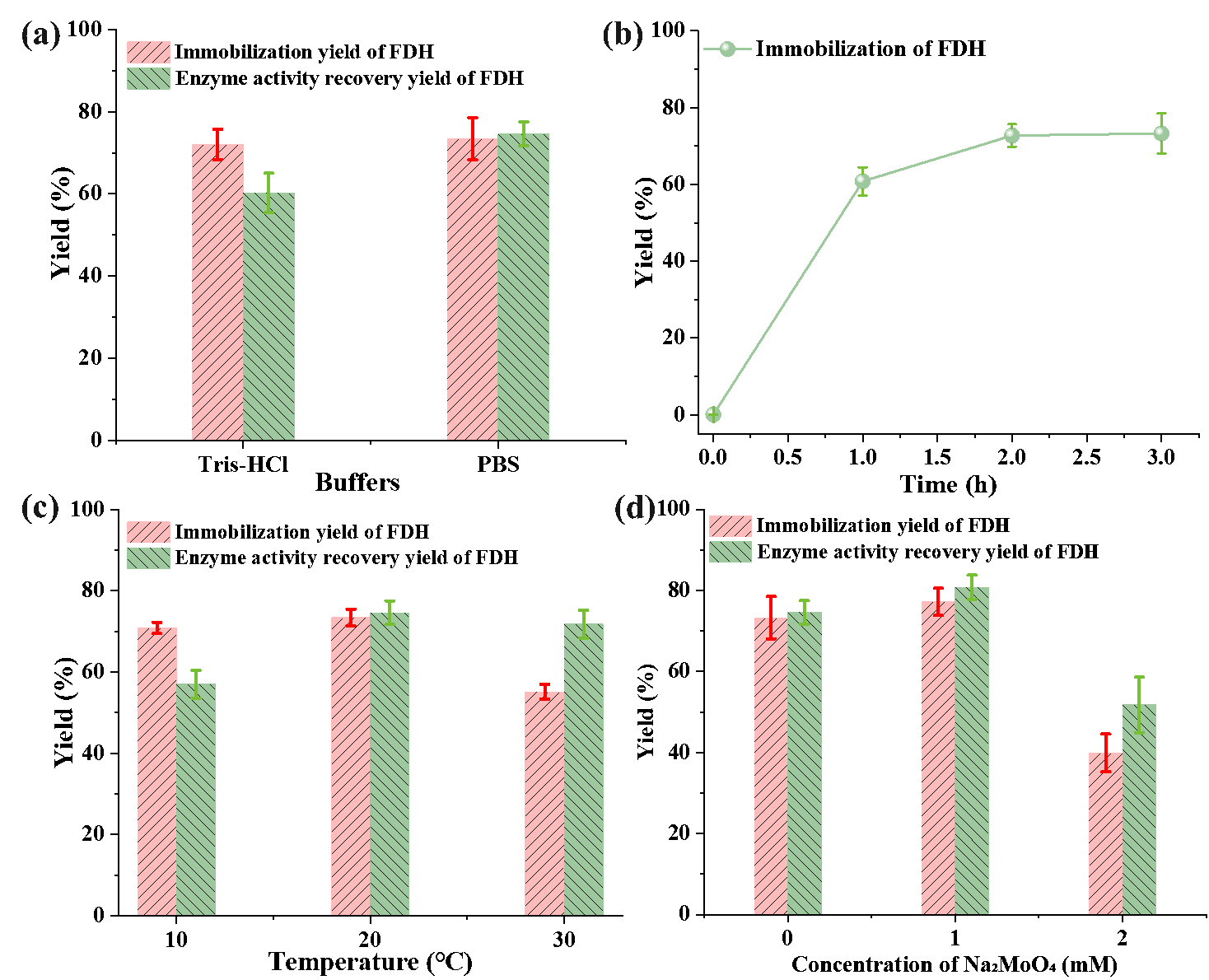
Since buffer composition is a critical factor influencing enzyme stability and activity [
32], we investigated the amount of formate dehydrogenase (FDH) immobilized by Au NCPs in two different buffer systems: Tris-HCl and PBS. as well as the enzyme activity recovery of the immobilized FDH. As illustrated in a, the immobilization yield of FDH by Au NCPs in both Tris-HCl and PBS buffer systems was similar; however, the enzyme activity recovery of the immobilized FDH in the PBS system was 14.35% higher than that observed in the Tris-HCl system. This finding suggests that PBS may provide a more favorable microenvironment for enzyme activity, likely due to its composition, which minimizes electrostatic interactions that could destabilize the enzyme structure. The positively charged Tris molecules may interfere with the negatively charged enzyme, reducing its bioactivity [
33,
34].
After establishing PBS as the preferred buffer, we further investigated the effect of reaction time on the FDH immobilization. As shown in b, the immobilization of FDH by the Au NCPs reached equilibrium after 2 h; the FDH immobilization yield was 72.74% at this time. Subsequently, we optimized the reaction temperature within the enzyme immobilization system. The immobilization yield of FDH by the Au NCPs and the enzyme activity recovery of the immobilized FDH initially increased and then decreased with rising reaction temperature. The highest immobilization rate of FDH (73.39%) and enzyme activity recovery (74.57%) were observed at a reaction temperature of 20 °C (c). However, at a reaction temperature of 30 °C, the enzyme activity recovery of the immobilized FDH decreased by only 3.77% compared to the 20 °C system. This indicates that the immobilized FDH maintained high enzyme activity at 30 °C, suggesting that the catalytic temperature of the immobilized FDH is well-suited for compatibility with the NADH regeneration system.
Molybdenum is a key component of the active sites of enzymes that catalyze essential redox reactions in carbon, nitrogen, and sulfur metabolism. Molybdenum ions are crucial for maintaining the stability and catalytic activity of FDH [
35,
36]. We further investigated the effect of sodium molybdate on the immobilization of FDH by the Au NCPs and the enzyme activity recovery of the immobilized FDH, as shown in d, both the immobilization yield and enzyme activity recovery of FDH initially increased and then decreased with increasing sodium molybdate concentration. When the concentration of sodium molybdate was 1 mM, the highest immobilization yield and enzyme activity recovery of the immobilized FDH were achieved by the Au NCPs, which were 77.19% and 80.16%, respectively. Whereas, when the sodium molybdate concentration was increased to 2 mM, the immobilization yield and enzyme activity recovery of FDH decreased by 37.29% and 29.06%, respectively. Therefore, the optimal conditions for FDH immobilization in Au NCPs were determined to be 2 h using PBS as the buffer, a controlled temperature of 20 °C, and 1 mM sodium molybdate as a stabilizer.
. (<b>a</b>) Effect of immobilization of FDH by buffer (buffer: pH = 7.40, 100 mM; Au NCPs: 1.50 mg/mL; FDH: 0.10 mg/mL; 20 °C; reaction time: 2 h); (<b>b</b>) The immobilization yield of FDH by Au NCPs as a function of reaction time (pH = 7.40, 100 mM PBS; Au NCPs: 1.50 mg/mL; FDH: 0.10 mg/mL; 20 °C); (<b>c</b>) The effect of temperature on the immobilization of FDH (pH = 7.40, 100 mM PBS; Au NCPs: 1.50 mg/mL; FDH: 0.10 mg/mL; reaction time: 2 h); (<b>d</b>) The effect of sodium molybdate on the immobilization of FDH (pH = 7.40, 100 mM PBS; Au NCPs: 1.50 mg/mL; FDH: 0.10 mg/mL; 20 °C; reaction time: 2 h).
The entry of CO
2 into solution is the primary rate-limiting step in enzyme-catalyzed carbon fixation processes [
37], and considering that the formation of CO
32− from CO
2 in water is thermodynamically spontaneous (CO
2 → CO
32−(aq)), with a standard Gibbs free-energy (Δ
fG
θ) of −527.8 kJ/mol [
38]. Previous studies have indicated that the Na
2CO
3 can effectively absorb CO
2 [
39,
40]. Therefore, we investigated the impact of varying Na
2CO
3 concentrations while introducing CO
2 (99.99%) into the reaction system to assess the formate concentration produced by the Au NCPs-FDH hybrid system under illuminated conditions. As illustrated in a, the reduction of CO
2 by the Au NCPs-FDH hybrid system in the absence of Na
2CO
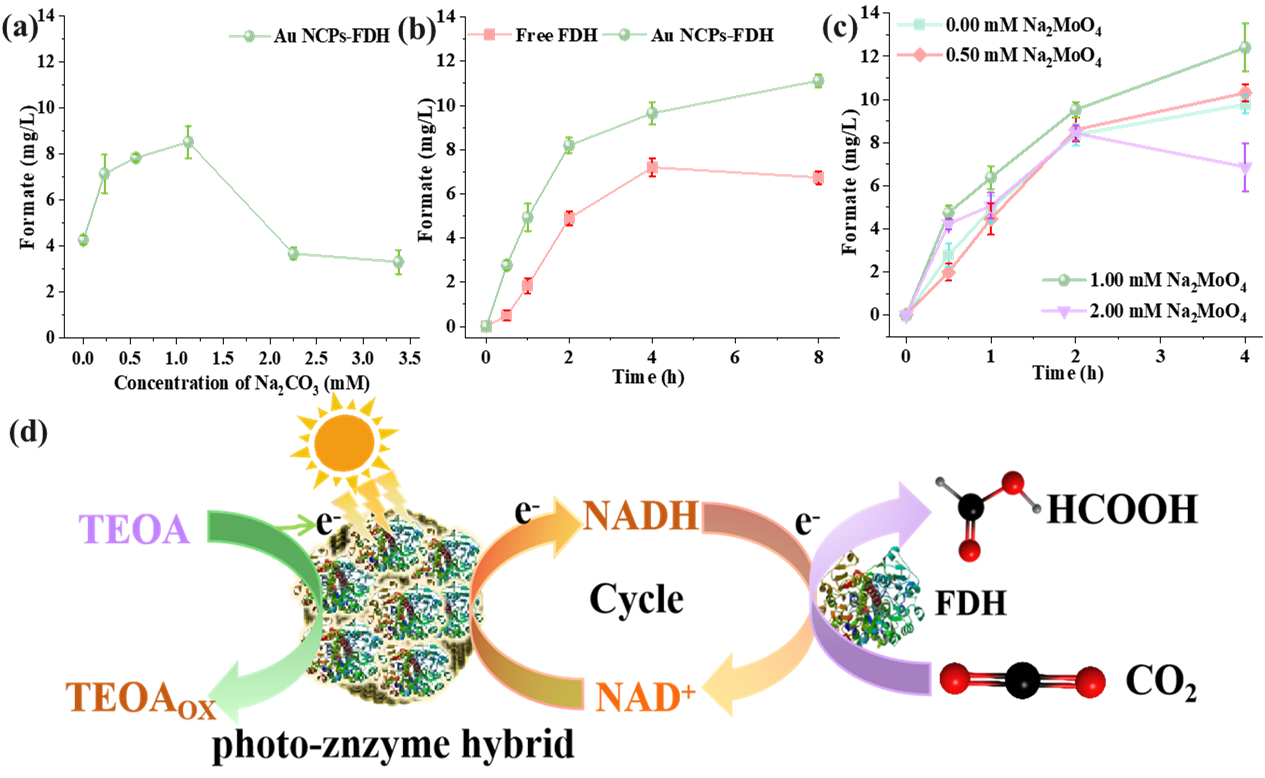
3 yielded only 4.27 mg/L of formate. However, increasing the concentration of Na
2CO
3 initially resulted in an increase in formate concentration, reaching a maximum of 1.13 mM Na
2CO
3, at which point the formate concentration was twice that of the system with 0 mM Na
2CO
3. This indicates that Na
2CO
3 enhances the reduction of CO
2 in the hybrid system, likely due to its capacity to absorb CO
2 and promote its dissolution in the liquid phase [
39]. Conversely, further increases in Na
2CO
3 concentration resulted in a rapid decline in formate levels to approximately 4 mg/L, comparable to the 0 mM Na
2CO
3 condition. This decrease may be attributed to a reduction in the bulk mass transfer coefficient of the solution at high Na
2CO
3 concentrations, which significantly lowers the CO
2 diffusion coefficient [
39], thereby resulting in reduced formate production.
We further investigated the performance of free FDH and the Au NCPs-FDH hybrid system in reducing CO
2 to produce formate under light exposure. The Au NCPs-FDH hybrid system demonstrated a higher formate synthesis rate (1.39 mg/L/h) compared to free FDH (0.84 mg/L/h). After 8 h of light exposure, the cumulative formate concentration in the Au NCPs-FDH hybrid system was 1.65 times greater than that of the free FDH system (b). These results indicate that the Au NCPs-FDH hybrid system effectively facilitates the light-driven reduction of CO
2 to formate.
. (<b>a</b>) Effect of sodium carbonate on the photocatalytic reduction of CO<sub>2</sub> to formate by Au NCPs-FDH hybrid (reaction time: 2 h); (<b>b</b>) Comparison of the performance of immobilized enzyme and free enzyme in reducing CO<sub>2</sub> under light (Na<sub>2</sub>CO<sub>3</sub>: 1.10 mM); (<b>c</b>) Effect of sodium molybdate on the photocatalytic reduction of CO<sub>2</sub> to formate by Au NCPs-FDH hybrid; (<b>d</b>) Schematic diagram of the mechanism for the continuous conversion of CO<sub>2</sub> to formate by Au NCPs-FDH hybrid system under Visible Light.
Furthermore, the addition of the FDH stabilizer sodium molybdate to the Au NCPs-FDH hybrid system, as illustrated in c, revealed that as the sodium molybdate concentration increased, the formate concentration of the hybrid system exhibited a pattern of initial increase and subsequent decrease. At a sodium molybdate concentration of 1 mM, the hybrid system achieved the highest formate concentration after 4 h of light exposure, reaching a concentration of 12.41 mg/L (269.61 µM). Additionally, the operation of the photo-enzyme hybrid system is illustrated in d. The Au NCPs act as a light absorber and charge generator. The photo-excited electrons generated by the Au NCPs directly reduce NAD
+ in situ to produce NADH, which then serves as a coenzyme for FDH, driving the conversion of CO
2 to formate. The released NAD
+ is further reduced by photoelectrons from the Au NCPs, enabling the cyclic regeneration of NADH. . Simultaneously, TEOA light oxidation replenishes the electrons consumed in the regeneration of NADH by the Au NCPs.
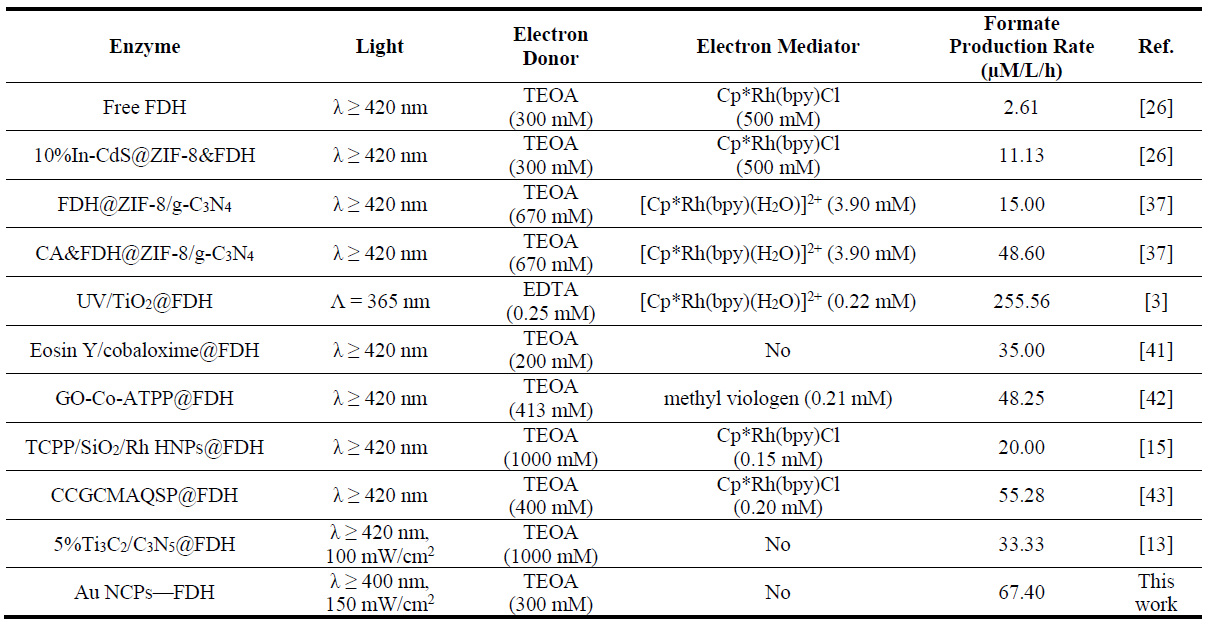
Moreover, the rate of formate production from CO
2 using the Au NCPs-FDH hybrid system under light exposure was compared with findings from other studies. The current system, which does not employ organic electron mediators, achieved a formate production rate of 67.40 µM/L/h with TEOA as the electron donor over 4 h of light exposure, obviously higher than most of the reported values (). This further demonstrates the efficiency and rapidity of the Au NCPs—FDH hybrid system in converting CO
2 to formate.
. Comparison of the performance of the photo-enzymatic conversion of CO2 to formate.
4. Conclusions
In conclusion, the photo-enzymatic hybrid system developed in this study, which integrates gold nanocapsule photocatalysts with formate dehydrogenase (FDH), successfully facilitated the light-driven in situ regeneration of NADH coenzymes for highly efficient catalytic CO2 reduction to produce formate. The reaction was conducted under irradiation with 150 mW/cm2 of white light, utilizing 300 mM TEOA as the electron donor, 1.10 mM Na2CO3 as a CO2 absorbent (supplemental carbon source), and 1 mM sodium molybdate as a stabilizer for FDH. Notably, this photo-enzymatic hybrid system achieved the highest formate concentration and production rate observed in this context, with a production rate that was 3.68 times greater than that of the free FDH-catalyzed system. This work demonstrates the feasibility of employing photo-enzymatic coupling for the continuous synthesis of formate from CO2, highlighting the potential for sustainable CO2 conversion technologies.
Author Contributions
Conceptualization, G.X. and H.S.; Methodology, Y.W.; Software, Y.W.; Validation, Y.W., M.G. and H.W.; Formal Analysis, Y.W.; Investigation, Y.W., M.G. and H.W.; Resources, J.X.; Data Curation, Y.W.; Writing—Original Draft Preparation, Y.W.; Writing—Review & Editing, G.X. and H.S.; Visualization, Y.W.; Supervision, H.S.; Project Administration, Y.W.; Funding Acquisition, G.X. and H.S.
Ethics Statement
Not applicable.
Informed Consent Statement
Not applicable.
Funding
This research was funded by the National Key R&D Program of China (Grant Nos. 2021YFC2103600) and the National Natural Science Foundation of China (Grant Nos. 22408378 and 22408019).
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
1.
Liu G, Wang L, Yan L, Zhao H, Li Y, Zhou L, et al.
A
dual-enzyme
microreactor
based
on
encapsulation
and
covalent
bond
for
enzymatic
electrocatalytic
CO
2 reduction.
Chem. Eng. J. 2023,
475, 146186.
[Google Scholar]
2.
Chai SYW, Ngu LH, How BS. Review
of
carbon
capture
absorbents
for
CO
2 utilization.
Greenh. Gases Sci. Technol. 2022,
12, 394–427.
[Google Scholar]
3.
Gu F, Wang Y, Meng Z, Liu W, Qiu L.
A
coupled
photocatalytic/enzymatic
system
for
sustainable
conversion
of
CO
2 to
formate.
Catal. Commun. 2020,
136, 105903.
[Google Scholar]
4.
Moon M, Park GW, Lee J-p, Lee J-S, Min K. Recent
progress
in
formate
dehydrogenase
(FDH)
as
a
non-photosynthetic
CO
2 utilizing
enzyme:
A
short
review.
J. CO2 Util. 2020,
42, 101353.
[Google Scholar]
5.
Liu W, Hou Y, Hou B, Zhao Z. Enzyme-catalyzed
sequential
reduction
of
carbon
dioxide
to
formaldehyde.
Chin. J. Chem. Eng. 2014,
22, 1328–1332.
[Google Scholar]
6.
Ji X, Su Z, Wang P, Ma G, Zhang S. Tethering
of
nicotinamide
adenine
dinucleotide
inside
hollow
nanofibers
for
high-yield
synthesis
of
methanol
from
carbon
dioxide
catalyzed
by
coencapsulated
multienzymes.
Acs Nano 2015,
9, 4600–4610.
[Google Scholar]
7.
Zhu D, Ao S, Deng H, Wang M, Qin C, Zhang J, et al.
Ordered
coimmobilization
of
a
multienzyme
cascade
system
with
a
metal
organic
framework
in
a
membrane:
Reduction
of
CO
2 to
methanol.
ACS Appl. Mater. Interfaces 2019,
11, 33581–33588.
[Google Scholar]
8.
Ryu J, Lee SH, Nam DH, Park CB. Rational
Design
and
Engineering
of
Quantum-Dot-Sensitized
TiO
2 Nanotube
Arrays
for
Artificial
Photosynthesis.
Adv. Mater. 2011,
16, 1883–1888.
[Google Scholar]
9.
Ji X, Su Z, Wang P, Ma G, Zhang S. Integration
of
artificial
photosynthesis
system
for
enhanced
electronic
energy-transfer
efficacy:
A
case
study
for
solar-energy
driven
bioconversion
of
carbon
dioxide
to
methanol.
Small 2016,
12, 4753–4762.
[Google Scholar]
10.
Huang X, Wang B, Wang Y, Jiang G, Feng J, Zhao H.
Photoenzymatic
enantioselective
intermolecular
radical
hydroalkylation.
Nature 2020,
584, 69–74.
[Google Scholar]
11.
Hutton GA, Reuillard B, Martindale BC, Caputo CA, Lockwood CW, Butt JN, et al.
Carbon
dots
as
versatile
photosensitizers
for
solar-driven
catalysis
with
redox
enzymes.
J. Am. Chem. Soc. 2016,
138, 16722–16730.
[Google Scholar]
12.
Guo M, Gu F, Meng L, Liao Q, Meng Z, Liu W. Synthesis
of
formaldehyde
from
CO
2 catalyzed
by
the
coupled
photo-enzyme
system.
Sep. Purif. Technol. 2022,
286, 120480.
[Google Scholar]
13.
Yang F, Zhang P, Qu J, Yang X, Cai Y, Li CM, et al. Highly
efficient
photoenzymatic
CO
2 reduction
dominated
by
2D/2D
MXene/C
3N
5 heterostructured
artificial
photosynthesis
platform.
J. Colloid Interface Sci. 2025,
678, 1121–1131.
[Google Scholar]
14.
Tan Y, Ma J, Zhang F, Wang S, Lan F, Liu H, et al.
Polymer
Photocatalyst
–Enzyme
Coupled
Artificial
Photosynthesis
System
for
CO
2 Reduction
into
Formate
Using
Water
as
the
Electron
Donor.
ACS Sustain. Chem. Eng. 2022,
10, 12065–12071.
[Google Scholar]
15.
Ji X, Wang J, Mei L, Tao W, Barrett A, Su Z, et al. Porphyrin/SiO
2/Cp*Rh(bpy)Cl
Hybrid
nanoparticles
mimicking
chloroplast
with
enhanced
electronic
energy
transfer
for
biocatalyzed
artificial
photosynthesis.
Adv. Funct. Mater. 2018,
28, 1705083.
[Google Scholar]
16.
Himiyama T, Waki M, Maegawa Y, Inagaki S. Cooperative
catalysis
of
an
alcohol
dehydrogenase
and
rhodium-modified
periodic
mesoporous
organosilica.
Angew. Chem. 2019,
131, 9248–9252.
[Google Scholar]
17.
Zhang P, Dong W, Zhang Y, Zhao L-N, Yuan H, Wang C, et al.
Metalated
carbon
nitride
with
facilitated
electron
transfer
pathway
for
selective
NADH
regeneration
and
photoenzyme-coupled
CO
2 reduction.
Chin. J. Catal. 2023,
54, 188–198.
[Google Scholar]
18.
Lian X, Fang Y, Joseph E, Wang Q, Li J, Banerjee S, et al. Enzyme
–MOF
(metal
–organic
framework)
composites.
Chem. Soc. Rev. 2017,
46, 3386–3401.
[Google Scholar]
19.
Zhang L, Can M, Ragsdale SW, Armstrong FA.
Fast
and
selective
photoreduction
of
CO
2 to
CO
catalyzed
by
a
complex
of
carbon
monoxide
dehydrogenase,
TiO
2,
and
Ag
nanoclusters.
ACS catal. 2018,
8, 2789–2795.
[Google Scholar]
20.
Liu D, Zhou F, Li C, Zhang T, Zhang H, Cai W, et al. Black
Gold:
Plasmonic
Colloidosomes
with
Broadband
Absorption
Self-Assembled
from
Monodispersed
Gold
Nanospheres
by
Using
a
Reverse
Emulsion
System.
Angew. Chem. Int. Ed. 2015,
54, 9596–9600.
[Google Scholar]
21.
Li X, Sun H, Mao X, Lao Y, Chen F. Enhanced
photosynthesis
of
carotenoids
in
microalgae
driven
by
light-harvesting
gold
nanoparticles.
ACS Sustain. Chem. Eng. 2020,
8, 7600–7608.
[Google Scholar]
22.
Wang Y, Jin Y, Wang Z, Xiao G, Su H. A
Light-Dark
Cascade
Procedure
for
the
Regeneration
of
NADH
using
Graphitic
Carbon
Nitride
Nanosheets.
ChemPhotoChem 2022,
6, e202200067.
[Google Scholar]
23.
Wu Y, Ward-Bond J, Li D, Zhang S, Shi J, Jiang Z.
g-C
3N
4@
α-Fe
2O
3/C
photocatalysts:
Synergistically
intensified
charge
generation
and
charge
transfer
for
NADH
regeneration.
ACS Catal. 2018,
8, 5664–5674.
[Google Scholar]
24.
Xu X-W, Zhang X-M, Liu C, Yang Y-L, Liu J-W, Cong H-P, et al. One-pot
colloidal
chemistry
route
to
homogeneous
and
doped
colloidosomes.
J. Am. Chem. Soc. 2013,
135, 12928–12931.
[Google Scholar]
25.
Song J, Cheng L, Liu A, Yin J, Kuang M, Duan H. Plasmonic
vesicles
of
amphiphilic
gold
nanocrystals:
Self-assembly
and
external-stimuli-triggered
destruction.
J. Am. Chem. Soc. 2011,
133, 10760–10763.
[Google Scholar]
26.
Zhou J, Tian X, Yu S, Zhao Z, Ji Y, Schwanebrg U, et al. In-CdS@
ZIF-8&FDH
photo-enzyme
nanosystem
with
high
NADH
regeneration
ability
via
indium
doping
for
enhanced
CO
2 reduction
to
formic
acid.
Chem. Eng. Sci. 2024,
285, 119613.
[Google Scholar]
27.
Wong C-H, Whitesides GM.
Enzyme-catalyzed
organic
synthesis:
NAD(P)H
cofactor
regeneration
by
using
glucose-6-phosphate
and
the
glucose-5-phosphate
dehydrogenase
from
Leuconostoc
mesenteroides.
J. Am. Chem. Soc. 1981,
103, 4890–4899.
[Google Scholar]
28.
Fang W, Deng Y, Tang L, Zeng G, Zhou Y, Xie X, et al. Synthesis
of
Pd/Au
bimetallic
nanoparticle-loaded
ultrathin
graphitic
carbon
nitride
nanosheets
for
highly
efficient
catalytic
reduction
of
p-nitrophenol.
J. Colloid Interface Sci. 2017,
490, 834–843.
[Google Scholar]
29.
Huang J, Antonietti M, Liu J.
Bio-inspired
carbon
nitride
mesoporous
spheres
for
artificial
photosynthesis:
Photocatalytic
cofactor
regeneration
for
sustainable
enzymatic
synthesis.
J. Mater. Chem. A 2014,
2, 7686–7693.
[Google Scholar]
30.
Cordas CM, Campaniço M, Baptista R, Maia LB, Moura I, Moura JJ. Direct
electrochemical
reduction
of
carbon
dioxide
by
a
molybdenum-containing
formate
dehydrogenase.
J. Inorg. Biochem. 2019,
196, 110694.
[Google Scholar]
31.
Dong G, Ryde U. Reaction
mechanism
of
formate
dehydrogenase
studied
by
computational
methods.
JBIC J. Biol. Inorg. Chem. 2018,
23, 1243–1254.
[Google Scholar]
32.
Wei T, Kaewtathip S, Shing K. Buffer
effect
on
protein
adsorption
at
liquid/solid
interface.
J. Phys. Chem. C 2009,
113, 2053–2062.
[Google Scholar]
33.
Sigurdardóttir SB, Lehmann J, Grivel JC, Zhang W, Kaiser A, Pinelo M. Alcohol
dehydrogenase
on
inorganic
powders:
Zeta
potential
and
particle
agglomeration
as
main
factors
determining
activity
during
immobilization.
Colloids Surf. B 2019,
175, 136–142.
[Google Scholar]
34.
Cugia F, Monduzzi M, Ninham BW, Salis A. Interplay
of
ion
specificity,
pH
and
buffers:
Insights
from
electrophoretic
mobility
and
pH
measurements
of
lysozyme
solutions.
RSC Adv. 2013,
3, 5882–5888.
[Google Scholar]
35.
Bulut H, Valjakka J, Yuksel B, Yilmazer B, Turunen O, Binay B. Effect
of
metal
ions
on
the
activity
of
ten
NAD-dependent
formate
dehydrogenases.
Protein J. 2020,
39, 519–530.
[Google Scholar]
36.
Maia LB, Moura JJ, Moura I.
Molybdenum
and
tungsten-dependent
formate
dehydrogenases.
BIC J. Biol. Inorg. Chem. 2015,
20, 287–309.
[Google Scholar]
37.
Yu S, Lv P, Xue P, Wang K, Yang Q, Zhou J, et al. Light-driven
enzymatic
nanosystem
for
highly
selective
production
of
formic
acid
from
CO
2.
Chem. Eng. J. 2021,
420, 127649.
[Google Scholar]
38.
Zheng Y, Zhang W, Li Y, Chen J, Yu B, Wang J, et al. Energy
related
CO
2 conversion
and
utilization:
Advanced
materials/nanomaterials,
reaction
mechanisms
and
technologies.
Nano Energy 2017,
40, 512–539.
[Google Scholar]
39.
Fang C, Zhang H, Xiao Y, Zhao T, Zou R, Luo G, et al. Carbon
dioxide
capture
in
sodium
carbonate
solution:
Mass
transfer
kinetics
and
DTAC
surfactant
enhancement
mechanism.
Carbon Capture Sci. Technol. 2024,
13, 100270.
[Google Scholar]
40.
Park SW, Sung DH, Choi BS, Oh KJ, Moon KH. Sorption
of
carbon
dioxide
onto
sodium
carbonate.
Sep. Sci. Technol. 2006,
41, 2665–2684.
[Google Scholar]
41.
Kim JA, Kim S, Lee J, Baeg J-O, Kim J. Photochemical
production
of
NADH
using
cobaloxime
catalysts
and
visible-light
energy.
Inorg. Chem. 2012,
51, 8057–8063.
[Google Scholar]
42.
Kumar S, Yadav RK, Ram K, Aguiar A, Koh J, Sobral AJ. Graphene
oxide
modified
cobalt
metallated
porphyrin
photocatalyst
for
conversion
of
formic
acid
from
carbon
dioxide.
J. CO2 Util. 2018,
27, 107–114.
[Google Scholar]
43.
Yadav RK, Baeg J-O, Oh GH, Park N-J, Kong K-j, Kim J, et al. A
photocatalyst
–enzyme
coupled
artificial
photosynthesis
system
for
solar
energy
in
production
of
formic
acid
from
CO
2.
J. Am. Chem. Soc. 2012,
134, 11455–11461.
[Google Scholar]