1. Introduction
As early as the 18th century, ammonia was used in agricultural production as a fertilizer. At present, the global annual production of nearly 200 million tons of ammonia is mainly used in agriculture and industry, and the demand is increasing year by year. Recently, ammonia is also used directly as a fuel or good carrier for hydrogen because its energy density is similar to fossil fuels and more stable than hydrogen energy. For example, the emergence of technologies such as ammonia fuel engines and NH
3 fuel cells highlights the significant potential of ammonia as a clean chemical energy source, thereby contributing to the achievement of carbon neutrality objectives [
1,
2,
3,
4]. However, with the increase in ammonia demand and application range, the environmental pollution caused by ammonia has become an urgent problem to be solved. Ammonia gas (NH
3) is a hazardous and malodorous pollutant released in substantial quantities from various sources [
5,
6], including agriculture [
7,
8], composting centers [
9], municipal biowaste [
10,
11,
12], vehicle exhaust [
13,
14,
15,
16], and fossil fuel combustion [
17]. NH
3 reacts with acidic gases in the air (such as NO
x and SO
2) to form particulate matter with a diameter of less than 2.5 µm (PM2.5) [
18,
19]. What’s more, NH
3 can generate secondary aerogel to promote the formation of haze and photochemical smog, causing serious pollution to the global atmospheric environment [
5]. Recent studies have shown that NH
3 emissions (25–39%) contributed more to PM2.5 than NO
x emissions (23–33%) globally in 2013 and suggest that controlling NH
3 emissions is much cheaper than controlling NO
x emissions [
20]. Moreover, NH
3 can irritate the human eye and respiratory organs and can be fatal in high concentrations, posing a serious threat to public health and safety. In order to protect people’s health, countries all over the world have strict regulations on NH
3 emissions. For example, the Occupational Safety and Health Administration sets the maximum exposure limit for NH
3 at 25 ppm for 8 h and the short-term exposure limit at 35 ppm for 15 min. That of the China Emission Standard for Odor Pollutants is less than 0.2 mg/m
3 (0.15 ppm). In addition, the researchers show that the cost of reducing NH
3 emissions is only 10% of the cost associated with controlling NO
x emissions, suggesting that managing NH
3 emissions can be a more economical approach to reducing both NO
x and PM2.5 emissions. If global NH
3 emissions are reduced by 50%, the implementation cost will be around $38 billion. This is significantly lower than the estimated social benefits of preventing deaths linked to NH
3 emissions and their contribution to PM2.5 pollution, which total $172 billion [
20]. Furthermore, the researchers further emphasize the priority of European policies aimed at controlling NH
3 emissions. For each unit of PM2.5 control, the cost−effectiveness of reducing NH
3 emissions is 5 to 11 times greater than that of reducing NO
x. Moreover, the impact of NH
3 reduction on nitrate and ammonium nitrogen levels is pronounced, with average annual sensitivities of 81.9% and 34.8%, respectively [
21]. Consequently, it is urgent to solve the NH
3 problem directly at the point source [
6].
At present, NH
3 emission has the characteristics of discrete emission, and the concentration is mostly ppm level. Conventional treatment strategies for NH
3 are divided into physical, biological and chemical methods. The physical methods include adsorption, absorption and condensation. Among them, the absorption and condensation methods are suitable for the treatment of high-concentration NH
3, but the equipment and operation costs are huge [
22,
23]. The adsorption method is suitable for the treatment of low and medium-concentrations of NH
3, but the change in the adsorbent and desorption process increases its cost [
24,
25]. The biological method also has the disadvantages of poor removal efficiency and large equipment footprint, and the culture and replacement of bacterial organisms will also cause huge costs [
26,
27]. In addition, thermal catalytic oxidation in chemical methods needs to be carried out at a higher temperature [
28,
29,
30,
31]. In contrast, photocatalytic oxidation has mild processing conditions and simplified equipment operation, providing a cost-effective and environmentally friendly route for NH
3 removal. Furthermore, this method has demonstrated tremendous potential in treating various gaseous pollutants, such as the removal of volatile organic compounds, purification of indoor formaldehyde, and reduction of NO
x emissions [
32,
33]. Therefore, it is very promising to remove NH
3 by direct photocatalytic oxidation to N
2 and H
2O at the point source. The design of efficient photocatalysts for NH₃ oxidation, capable of achieving high activity in NH₃ conversion while minimizing the formation of by-products such as NOₓ and N₂O, represents one of the core challenges in this field. In this case, it is necessary to select an economical and efficient photocatalyst with high nitrogen selectivity. As a common photocatalyst, TiO
2 has been widely used in the photocatalytic removal of NH
3. Under the irradiation of light, the electrons in the valence band (VB) of TiO
2 are excited to transition to the conduction band (CB) and form a photogenerated carrier (electron-hole pair). Subsequently, part of the photogenerated charge carriers (
e− and
h+) migrated to the surface of TiO
2 and reacted with the adsorbed substances on the surface or produced active substances (
·O
2− and
·OH) [
34]. The active substance will also be further involved in the chemical reaction, thus oxidizing the NH
3 to N
2 and H
2O. However, the application of TiO
2 is restricted by its wide band gap width and low carrier utilization [
34,
35,
36]. To address these limitations, the researchers have employed strategies such as surface modification, metal loading, doping modification, and semiconductor compositing to enhance the utilization of solar energy and carriers in TiO
2 [
37,
38,
39,
40]. Additionally, the impact of different modification strategies on the photocatalytic activity of catalysts and the mechanism of NH
3 removal is still not clear.
In this review, we aim to provide guidance for the development of NH
3 photocatalytic oxidation systems. Relevant researches on photocatalytic oxidation of NH
3 in recent years are analyzed, with a focus on photocatalytic oxidation performance, carrier and solar energy utilization efficiency, as well as the oxidation pathway of NH
3. The purpose of this paper is to clarify the difficulties and challenges existing in the current photocatalytic oxidation of NH
3, and put forward our views on improving the photocatalytic efficiency of NH
3, so as to provide new ideas and guidance for the removal of environmental pollutants.
2. Catalyst for Photocatalytic Oxidation of NH3
2.1. TiO2 Based Photocatalyst
TiO
2 has emerged as an advanced photocatalyst for tackling environmental pollution due to its eco-friendliness, low cost, renewability, and high photocatalytic activity. However, the wide bandgap of TiO
2 results in low efficiency in utilizing visible light and significant carrier recombination, which leads to suboptimal photocatalytic performance [
34]. Nevertheless, a range of modification techniques, such as doping with metal or non-metal ions, loading metal co-catalysts, and constructing heterostructured semiconductor catalysts, can greatly improve its efficiency in visible light utilization and carrier separation. In this section, we first studied the application of TiO
2-based photocatalyst in the photocatalytic oxidation of NH
3.
2.1.1. Pure TiO
2 Photocatalyst
TiO
2 has three common crystal structures, among which anatase and rutile are the two most studied polymorphs. The bandgap width of rutile is approximately 3.0 eV, while that of anatase is about 3.2 eV. From the perspective of bandgap width, rutile TiO
2 has a narrower bandgap, which implies a broader range of light absorption [
41]. However, the recombination of charge carriers in rutile TiO
2 is more severe, resulting in its photocatalytic ability being inferior to that of anatase TiO
2. Additionally, anatase TiO
2 has a stronger hydroxyl acidity on its surface, leading to a greater adsorption capacity for NH
3 molecules. Wu et al. prepared a modified TiO
2 catalyst using the sol−gel method and tested it under visible and ultraviolet light (Figure 1a). The results showed that the TiO
2 catalyst calcined at 400 °C exhibited the best ultraviolet light activity with a conversion rate of approximately 43%. Their study found that surface acidic sites play a crucial role in NH
3 PCO. As the calcination temperature increased, the specific surface area and pore volume of the TiO
2 samples gradually decreased. However, the sample calcined at 400 °C still maintained a relatively high specific surface area (145.2 m²/g) and good crystallinity. The high specific surface area and good crystallinity are conducive to forming more acidic sites. Additionally, after high-temperature calcination, surface carbon impurities were reduced, thereby preserving more acidic sites [
42].
Although anatase offers distinct advantages, TiO
2 containing both anatase and rutile phases (such as P25) is widely used in various catalytic reactions. Heylen et al. show that the conversion efficiency of commercial P25 for NH
3 at 150 °C is only 60%, primarily yielding by-products like NO and NO
2, with an N
2 selectivity of merely 28%. Even when the space velocity is reduced by half, the selectivity of P25 does not significantly improve. In contrast, the PC500 TiO
2 catalyst achieves a 99% conversion rate and 92% dinitrogen selectivity under conditions of 150 °C, 300 VHSV h⁻¹, and 1.1 mW/cm² UVA irradiation. Compared to P25 and UV100, PC500 exhibits greater weight loss (approximately 12%) in thermogravimetric analysis (TGA), indicating a higher water adsorption capacity on its surface, which may enhance its NH
3 adsorption capability. At lower temperatures (100 and 50 °C), the NH
3 conversion rate of PC500 decreases, but the selectivity for NO increases, suggesting the potential involvement of an internal selective catalytic reduction (iSCR) mechanism [
43]. In addition, Sola et al. compared the differences in the gas-phase photocatalytic degradation of NH
3 and ethanol on two commercial TiO
2 materials (Evonik’s P25 and Sigma Aldrich (SA)) in the presence of water. The photodegradation process was tracked using FTIR to analyze changes over irradiation time. The research found that during the NH
3 photodegradation process, surface nitrate species remained firmly bound on P25, while they were eliminated on SA. This was attributed to the presence of more and stronger Lewis acid sites on the SA surface, as well as a greater number of reactive surface hydroxyl species. The adsorbed NH
3 could react with the generated nitrate species to form N
2, a reaction mechanism that was lacking on the P25 surface (Figure 1b) [
44]. In summary, for pure TiO
2 photocatalysts, the anatase phase seems to be more suitable for the photocatalytic oxidation of NH
3 because of its better removal performance and N
2 selectivity than the rutile phase.
In addition, the main exposed crystal plane of anatase TiO
2 is the (1 0 1) crystal plane because its average surface free energy (0.44 J/m
2) is lower than that of the (0 0 1) crystal plane (0.90 J/m
2). However, He et al. prepared TiO
2 with the main exposure surface of (0 0 1) crystal plane by surface fluoridation with HF acid and found that it had better performance for the photocatalytic oxidation of NH
3. The experimental results (Figure 1c) show that the ≡Ti−F group on the surface of the photocatalyst can delay the recombination of photogenerated
e− and
h+, which may be the main reason for the excellent activity of F−TiO
2 catalyst [
45]. Further, using H
2O and NH
3 photooxidation as probe reactions, they investigated the effect of surface F ions on carrier migration in anatase TiO
2 crystals dominated by (0 0 1) or (1 0 1) planes. Fluorinated (F-T001) and defluorinated (T001) TiO
2 with exposed (001) crystal faces and TiO
2 with exposed (1 0 1) crystal faces (T101) and fluorinated (1 0 1) (T101-F) and (0 0 1) (T001-F) crystal faces are synthesized. They observed a significant synergistic effect of the (0 0 1) plane and surface F ions on photogenic hole migration. The (0 0 1) plane provides
h+ trapping sites, and the electrostatic effect of surface F ions attracts and accelerates the migration of
h+ to the (0 0 1) plane and collaboratively promotes the separation of electron-hole pairs, thus significantly improving the photooxidation activity (Figure 1d). However, there is no synergy between the (1 0 1) plane and the surface F ions [
46]. Furthermore, they prepared anatase TiO
2 with (0 0 1), (1 0 1) and (0 1 0) clean dominant planes (denoted as T001, T101 and T010) and tested their photocatalytic activity for NH
3 oxidation. The results (Figure 1e) showed that the order of photocatalytic activity of NH
3 oxidation is (0 0 1) > (1 0 1) > (0 1 0). This is due to the special effect of the (0 0 1) crystal plane on the
h+, which can promote the transition of NH
3 molecules to·NH
2 [
47].
Figure 1. (<strong>a</strong>) NH<sub>3</sub> photocatalytic oxidation of TiO<sub>2</sub> at different calcination temperatures [
42]. (<strong>b</strong>) The variation of different species on P25 and anatase TiO<sub>2</sub> over time [
44]. (<strong>c</strong>) The photocatalytic oxidation of NH<sub>3</sub> on surface-fated TiO<sub>2</sub> (F−TiO<sub>2</sub>), defluorinated TiO<sub>2</sub> (D−TiO<sub>2</sub>) and pure TiO<sub>2</sub> [
45]. (<strong>d</strong>) IR spectra of F-T001, T001, T001-F, T101-F and T101 after 5 min UV irradiation under N<sub>2</sub> atmosphere [
46]. (<strong>e</strong>) The photocatalytic oxidation of NH<sub>3</sub> on T010, T101 and T001 [
47]. (<strong>f</strong>) Synergistic removal of NH<sub>3</sub> and butyraldehyde by DBD and photocatalytic oxidation on TiO<sub>2</sub> [
48]. Reproduced with permission. Copyright 2014, 2016, 2017 and 2018, Elsevier Publication.
Several studies have investigated the efficacy of TiO
2 photocatalysis in the removal of NH
3 from mixed gas pollutants under practical conditions. Research conducted by Saoud et al. demonstrates that when pollutants are treated individually, the removal efficiency for the removal of NH
3 is 15%, while that for butanal is 45%. However, when both pollutants are treated simultaneously, the removal efficiency for NH
3 decreases to 12%, and for butanal, it decreases to 33%. This reduction in photocatalytic oxidation efficiency is likely due to the competitive adsorption of the two pollutants on the active sites. In contrast, dielectric barrier discharge (DBD) plasma exhibits a higher degradation efficiency for ammonia (Figure 1f). Under specific energy input (SEI) conditions, DBD plasma alone achieves a removal efficiency of 51%. Furthermore, coupling DBD plasma with photocatalytic oxidation significantly enhances the removal efficiency for ammonia, achieving a rate of 83%, indicating a synergistic interaction between the two methods [
48].
2.1.2. Doped Modified TiO
2 Photocatalyst
Doping modification is mainly divided into metal element doping (Fe, Cu, Ce,
etc.), non-metallic element doping (N, S, C,
etc.) and metal and non-metallic element co-doping. For the photocatalytic oxidation of ammonia, doping modification mainly contributes to three aspects. (i) The formation of oxygen vacancy (Ov). On the one hand, Ov, as a Lewis acidic site, can promote the adsorption of NH
3. On the other hand, Ov can also be used as
e−−
h+ traps to promote the separation of photogenerated carriers and enhance photocatalytic activity. (ii) As an
e−−
h+ trap. Similar to the role of Ov, the introduced ions can also act as electron or hole traps, thereby improving carrier utilization. (iii) Broaden the optical response range. The introduced elements can form impurity levels below the CB or above the VB of the photocatalytic semiconductor, thereby reducing the band gap (
Eg) width of the photocatalyst and broadening the light absorption range. However, a significant drawback of doping is that the introduction of heteroatoms, whether through atomic substitution or occupancy of interstitial sites, inevitably alters the atomic structure of the photocatalyst and may lead to the formation of recombination centers [
49]. Excessive doping can diminish catalytic performance by modifying the atomic structure and creating additional recombination sites. Therefore, precise optimization of the doping ratio is essential for effective modification of the photocatalyst.
Wang et al. prepared Fe-doped TiO
2 (Fe−TiO
2) thin films by sol−gel method (Figure 2a) to degrade typical indoor air pollutants HCHO, NH
3 and C
6H
6 under sunlight irradiation, and the removal efficiency of NH
3 was 53.1% after 9 h. By replacing Ti
4+ ion, Fe
3+ ion introduced a new impurity energy level between the CB and the VB, which decreased the E
g and inhibited the
e−−
h+ pair recombination and improved the photocatalytic activity [
50]. In addition, the researchers prepared 5% N/Ag−TiO
2 by in-situ solvothermal method, which showed 4.3 times higher photocatalytic performance than pure TiO
2. The introduction of Ag ions formed an impurity energy level below the CB, which significantly reduced the band gap energy of TiO
2 from 3.2 to 1.7 eV and increased the absorption of visible light (Figure 2b). At the same time, Ag ion acted as an
e− trapping site and slowed down the recombination of carriers. N doping increases the specific surface area of the catalyst and enhances the NH
3 adsorption [
51]. Similarly, Wang et al. synthesized Mo, C−TiO
2 photocatalyst by sol−gel synthesis and low-temperature calcination using organic groups in TiO
2 precursor as a carbon source. Experimental results demonstrated that the incorporation of carbon extended the photo-response range of TiO
2, while the inclusion of Mo was found to effectively suppress the recombination of photogenerated carriers under both UV and visible light conditions (Figure 2c). The co-doping of Mo and C further facilitated the formation of Ov, thereby improving the photocatalytic oxidation performance under both UV and visible light irradiation. Meanwhile, in their proposed N
2H
4 mechanism, Mo ions served as the reactive active site, and NH
3 adsorbed on it reacted with the reactive oxygen species gradually to generate N
2 as the expected end product [
52].
In addition, non-metallic elements (such as C, N) are also used to improve the photocatalytic NH
3 removal performance of TiO
2. Using urea as the N source, Jiang et al. prepared N−TiO
2 and used it for NH
3 removal under visible light. The results show (Figure 2d) that the efficiency of N−TiO
2 photocatalytic oxidation of NH
3 remains above 80%, and the N
2 selectivity is stable at about 87%. In contrast, the NH
3 conversion on pure TiO
2 stabilized at about 67% within 72 h, and the nitrogen selectivity did not exceed 55%. N element was successfully doped into the TiO
2 lattice system in the form of O−Ti−N, which reduced the band gap energy from 3.18 to 3.07 eV, improving the light utilization rate. Secondly, the water molecules formed during the reaction contribute to the continuous formation of hydroxyl radical (·OH) in the in-situ reaction, thus improving the stability of the reaction [
53]. Further, Gao et al. added a carbon layer to N−TiO
2 and tested the activity of NH
3 photocatalytic oxidation under visible light (Figure 2e). The results show that the ammonia removal efficiency and N
2 selectivity of C/N−TiO
2 catalyst is 94% and 98%, respectively. The co-doping of C and N reduces the band gap of TiO
2 (3.18~2.90 eV), thus broadening the photo-response range of TiO
2. At the same time, more Ov was formed under visible light irradiation, which improved the activity of the catalyst. Additionally, more acidic sites were introduced, which improved the adsorption capacity of the catalyst for NH
3 [
54].
Figure 2. (<strong>a</strong>) Kinetics of degradation reaction of the mixed pollutants on Fe−TiO<sub>2</sub> [
50]. Reproduced with permission. Copyright 2014, WILEY Publication. (<strong>b</strong>) Schematic diagrams of the band energy structure and the migration of photoexcited electrons in undoped TiO<sub>2</sub>, N−TiO<sub>2</sub>, Ag−TiO<sub>2</sub>, and N/Ag−TiO<sub>2</sub> [
51]. Reproduced with permission. Copyright 2020, MDPI Publication. (<strong>c</strong>) Schematic diagram of photocatalytic oxidation of NH<sub>3</sub> on Mo, C−TiO<sub>2</sub> [
52]. (<strong>d</strong>,<strong>e</strong>) Photocatalytic oxidation of NH<sub>3</sub> on N−TiO<sub>2</sub>, C/N−TiO<sub>2</sub> and pure TiO<sub>2</sub> under visible light [
53,
54]. Reproduced with permission. Copyright 2023 and 2024, Elsevier Publication.
2.1.3. Semiconductor Composite Modified TiO
2
Coupling TiO
2 with other semiconductors to construct heterostructures can greatly facilitate charge transfer between interfaces and promote the separation and migration of photogenerated charges, thus improving carrier utilization. Currently, the common types of heterojunctions are type-I, type-II, Z-Scheme and S-Scheme.
Cu-based semiconductor (CuO and Cu
2O), as a common visible light photocatalyst, is a good choice for TiO
2 composite due to their narrow band gap. The researchers prepared Cu
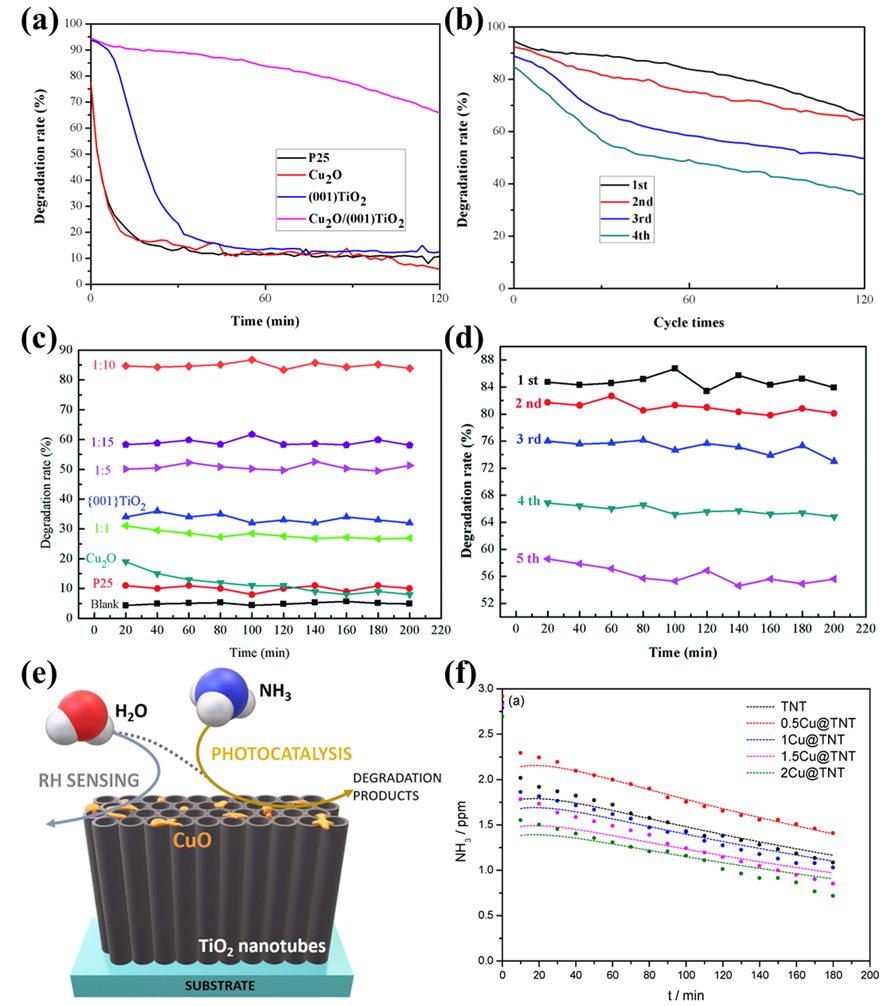
2O/(0 0 1) TiO
2 by an impregnation-reduction method and tested the removal efficiency of NH
3 under simulated sunlight. The results (Figure 3a,b) show that the removal rate of NH
3 by Cu
2O/(0 0 1)TiO
2 is 80% (The NH
3 concentration is about 120 ppm), which is higher than that of pure P25 (12%), Cu
2O (12%) and (0 0 1) TiO
2 (15%). This is due to the fact that Cu
2O promotes the exposure of the (0 0 1) TiO
2 active crystal plane and broadens the absorption range of sunlight. Further, after repeated use for 4 times, the degradation rate of NH
3 gradually decreased, which was caused by the formation of nitrates covering the reactive site [
55]. Further, Zhu et al. explored the effect of the composite ratio of Cu
2O and (0 0 1) TiO
2 on the photocatalytic removal of NH
3. The results show (Figure 3c,d) that when the recombination ratio was 1:10, the specific surface area was the largest (72.51 m
2/g) and the degradation rate of NH
3 was also maintained at 85%. However, after cycling, the catalyst was still significantly deactivated, and it was found that the surface adsorbed water and hydroxyl radical participated in the oxidation of NH
3, and finally formed nitrate species [
56]. Additionally, Tihana et al. prepared Cu-modified TiO
2 nanotube arrays by wet impregnation (Figure 3e) and anodic oxidation, which have the dual roles of photocatalytic NH
3 degradation and as relative humidity sensing materials. Compared with unmodified TNT, all CuO-TNTs exhibited better NH
3 removal efficiency (Figure 3f). The CuO nanoparticles dispersed on the TiO
2 surface acted as free electron trapping sites, which reduced the rapid recombination rate of electrons and holes on the TiO
2 surface, thus promoting the photocatalytic degradation of NH
3. The above composites with Cu-based photocatalysts are anatase TiO
2, but special results are obtained when they are combined with rutile TiO
2 [
57]. Chen et al. investigated the effect of charge transfer direction between CuO
x and TiO
2 (Figure 4a) on the photocatalytic degradation activity of NH
3. They observed that CuO
x/anatase TiO
2 exhibited better catalytic activity than CuO
x/rutile TiO
2 under UV irradiation. Whereas CuO
x/rutile TiO
2 had excellent photocatalytic activity under visible light irradiation, CuO
x/anatase TiO
2 was inactive. Combined with EPR and DFT calculations, they found that the crystalline phase of TiO
2 significantly affects the charge separation of CuO
x/TiO
2, leading to different charge transfer directions (Figure 4b,c). Visible light can excite electrons from the VB of rutile to CuO
x, leaving behind
h+ with high oxidation potentials that effectively oxidize NH
3, whereas at the CuO
x/anatase TiO
2 interface, visible light can only excite the electron transfer from the VB of CuO
x to the CB, and the
h+ left behind in its VB cannot oxidize NH
3 with low oxidation potentials [
58].
Figure 3. (<strong>a</strong>,<strong>b</strong>) The photocatalytic oxidation of NH<sub>3</sub> on P25, Cu<sub>2</sub>O, (0 0 1)TiO<sub>2</sub> and Cu<sub>2</sub>O/(0 0 1)TiO<sub>2</sub> and the NH<sub>3</sub> removal efficiency after cycling on Cu<sub>2</sub>O/(0 0 1)TiO<sub>2</sub> under visible light [
55]. Reproduced with permission. Copyright 2019, MDPI Publication. (<strong>c</strong>,<strong>d</strong>) Photocatalytic oxidation of ammonia at different Cu<sub>2</sub>O and (0 0 1) TiO<sub>2</sub> composite ratios and removal efficiency of NH<sub>3</sub> after cycling when Cu<sub>2</sub>O (0 0 1) is 1:10 [
56]. Reproduced with permission. Copyright 2021, RSC Publication. (<strong>e</strong>,<strong>f</strong>) Photocatalytic oxidation of NH<sub>3</sub> on N−TiO<sub>2</sub>, C/N−TiO<sub>2</sub> and pure TiO<sub>2</sub> under visible light [
57]. Reproduced with permission. Copyright 2021, MDPI Publication.
Figure 4. (<strong>a</strong>) Photocatalytic oxidation of NH<sub>3</sub> under visible light over CuO<sub>x</sub>/anatase TiO<sub>2</sub> and CuO<sub>x</sub>/rutile TiO<sub>2</sub>. (100 ppm NH<sub>3</sub>, 20 vol% O<sub>2</sub>, RH = 50% and N<sub>2</sub> balance). (<strong>b</strong>) EPR spectra of CuO<sub>x</sub>/rutile TiO<sub>2</sub> and CuO<sub>x</sub>/anatase TiO<sub>2</sub> measured at 77 K under vacuum with visible light or UV irradiation. (<strong>c</strong>) DMPO spin-trapping EPR spectra measured at 303 K after 5 min visible irradiation in aqueous solutions and acetonitrile over different samples [
58]. Reproduced with permission. Copyright 2022, Elsevier Publication.
In addition to Cu-based catalysts, other semiconductors have also been used to compound with TiO
2 to enhance the photocatalytic oxidation activity of NH
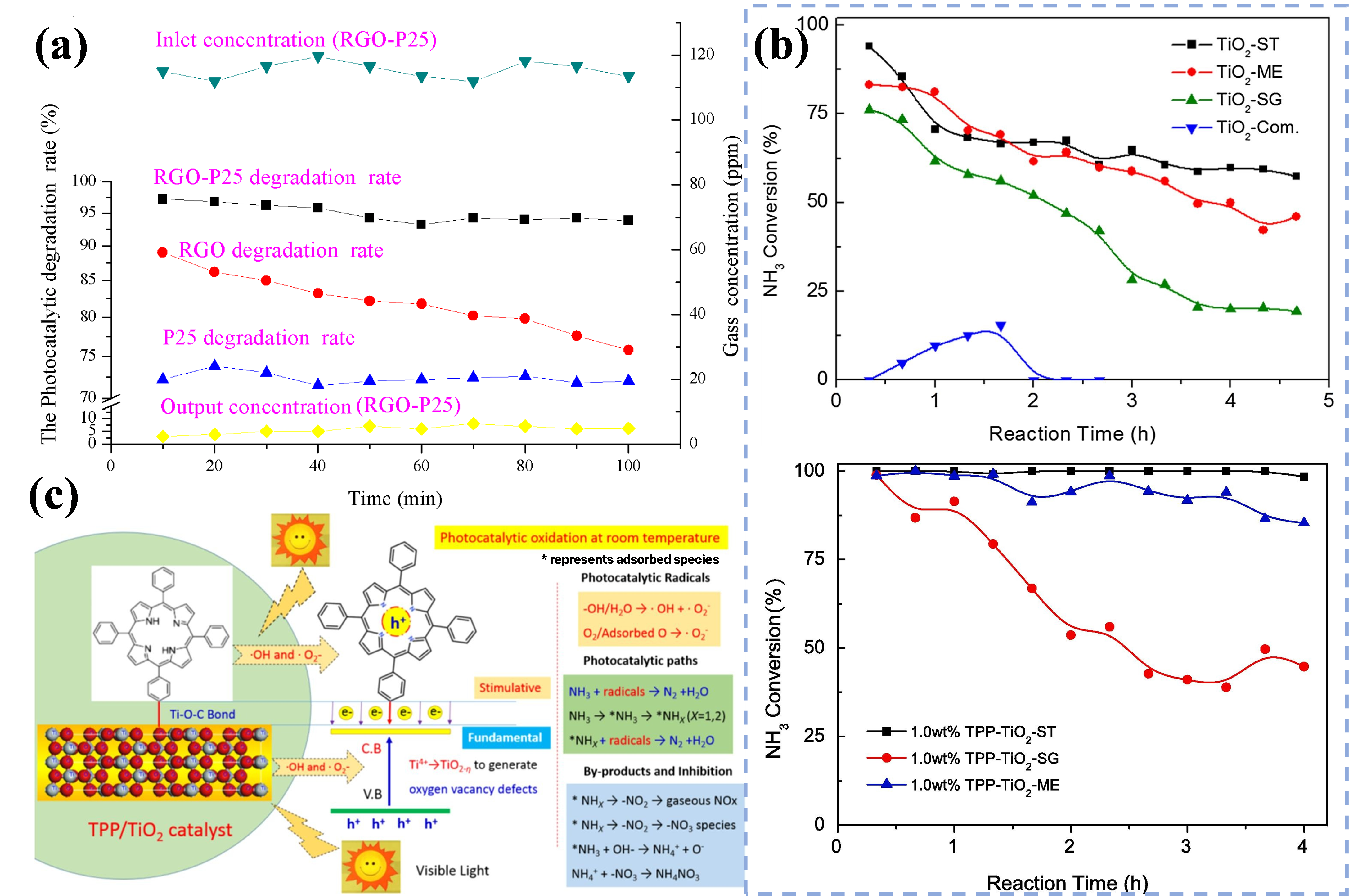
3. To broaden the sunlight response range, Pu et al. prepared a composite RGO/TiO
2 catalyst for NH
3 degradation in livestock farms by Hummer’s method using a silica core board as a carrier. The degradation efficiency of NH
3 was as high as 93.64% under 300 W UV irradiation (Figure 5a). The introduction of RGO reduced the band gap energy of P25 from 3.14 to 2.96 eV, which enhanced the light absorption. Meanwhile, its layered pleated structure increased the adsorption of NH
3 on the surface of the material and also lowered the recombination rate of photogenerated carriers, which led to the improvement of the photocatalytic performance of P25 [
59]. Further, Gao et al. prepared tetraphenyl porphyrin (TPP)-modified anatase TiO
2 by a one-step solvothermal method, which exhibited excellent photocatalytic activity under visible light irradiation, and the ammonia removal efficiency reached more than 98% within 8 h at room temperature. Under visible light irradiation, TPP contributes H to generate H
+ and VB holes with strong oxidizing ability (Figure 5b). The electrons were transferred to the TiO
2 surface via Ti−O−C bonds, which promoted the reduction of Ti
4+ and the generation of oxygen vacancy defects. However, the accumulation of thermodynamically stable NH
4NO
3 (Figure 5c) on the catalyst surface leads to the deactivation of the photocatalyst [
60].
Figure 5. (<strong>a</strong>) The degradation effect of NH<sub>3</sub> by RGO-P25, RGO and P25 [
59]. Reproduced with permission. Copyright 2018, MDPI Publication. (<strong>b</strong>) Photocatalytic activity of ammonia oxidation on different catalysts (100 ppm NH<sub>3</sub>, 21 vol% O<sub>2</sub>, N<sub>2</sub> to balance, the total flow rate of 0.2 L/min, visible light area of 47.25 cm<sup>2</sup>, light intensity of 100 mw/cm<sup>2</sup>). (<strong>c</strong>) Proposed photocatalytic oxidation mechanisms of NH<sub>3</sub> over TPP/TiO<sub>2</sub>−ST catalysts under visible light irradiation [
60]. Reproduced with permission. Copyright 2020, Elsevier Publication.
To enhance the capture of NH
3, the researchers also used a semiconductor coating on another photocatalyst to increase the specific surface area of the catalyst. Pu et al. prepared MoS
2@TiO
2 encapsulated carbon coaxial nanobelts (CNBs) by electrostatic spinning-hydrothermal reaction (Figure 6a) for photocatalytic degradation of NH
3. The degradation efficiency reached 91% after only 7 min of UV illumination. The introduction of carbon improved the carrier dynamics and electron affinity of TiO
2, and the larger specific surface area and layered pore structure of MoS
2@TiO
2 CNBs enhanced the adsorption of NH
3. While the construction of heterojunctions promoted the separation and migration of photogenerated charges, effectively reduced the recombination efficiency, and prolonged the lifetime of photogenerated
e−−
h+ [
61]. Similarly, Li et al. used the sol−gel method to coat TiO
2 on WO
3 nanowires (Figure 6b). After the WO
3@TiO
2 photocatalyst was exposed to simulated sunlight for 3 h, the NH
3 conversion and N
2 selectivity were found to be 58% and 94%, which were approximately double the performance of TiO
2 and WO
3 alone (Figure 6c). The enhanced photocatalytic activity is attributed to the built-in electric field between WO
3 and TiO
2, which promotes rapid charge separation and migration. In addition, the core-shell structure enhanced NH
3 adsorption and O
2 activation. This study suggests that photogenerated holes and superoxide radicals play important roles in the photocatalytic oxidation of NH
3, and efficient removal of NH
3 by constructing an interfacial electric field is an effective strategy [
62]. In addition, Li et al. prepared a porous carbon framework material with a layered structure of N-doped coated ultrafine TiO
2 (N−C@TiO
2) powder by solvothermal method, which has excellent photocatalytic activity, and the removal rate of NH
3 reaches 100% after only 5 min of illumination. The N-doped porous carbon framework has a large specific surface area and good metal conductivity, which can promote the charge transfer to the interface and impede the photogenerated
e−−
h+ recombination. The increase in the concentration of
h+ on the TiO
2 surface can promote the adsorption and activation of NH
3, thus enhancing the photocatalytic activity of TiO
2 [
63].
Figure 6. (<strong>a</strong>) Schematic illustration of the proposed photocurrent−transducer mechanism and the NH<sub>3</sub> degradation of P25, TiO<sub>2</sub> CNBs and MoS<sub>2</sub>@TiO<sub>2</sub> CNBs heterojunction [
61]. (<strong>b</strong>) Schematic illustration of the photocatalytic NH<sub>3</sub> oxidation mechanism over the 3WO<sub>3</sub>@TiO<sub>2</sub> photocatalyst under simulated sunlight irradiation [
62]. (<strong>c</strong>) Schematic illustration of the hierarchical structure of as−synthesized material (N−C@TiO<sub>2</sub>) and involved electron behavior for photodegradation NH<sub>3</sub> [
63]. Reproduced with permission. Copyright 2018, 2020 and 2024, Elsevier Publication.
For the photocatalytic oxidation of NH
3, at present, it is limited to the formation of type-II heterostructures to improve the utilization of carrier and solar energy. However, the type II heterostructure is at the expense of the Oxidation and reduction ability of the photocatalyst. Therefore, it is necessary to develop other heterogeneous structures, especially those with Z-type heterogeneous structures.
2.1.4. Metal-supported Modified TiO
2
Depositing different noble metals on the TiO
2 surface, the photogenerated electrons will continue to be transferred from TiO
2 to the metal due to the difference in their Fermi energy levels until their Fermi energy levels are similar or the same. The Schottky junction formed at the interface between the metal and TiO
2 can promote the separation of photogenerated charges. The metal acts as an electron trapping site to enrich electrons and reduce O
2 adsorbed on the surface to generate more reactive species, delaying carrier recombination.
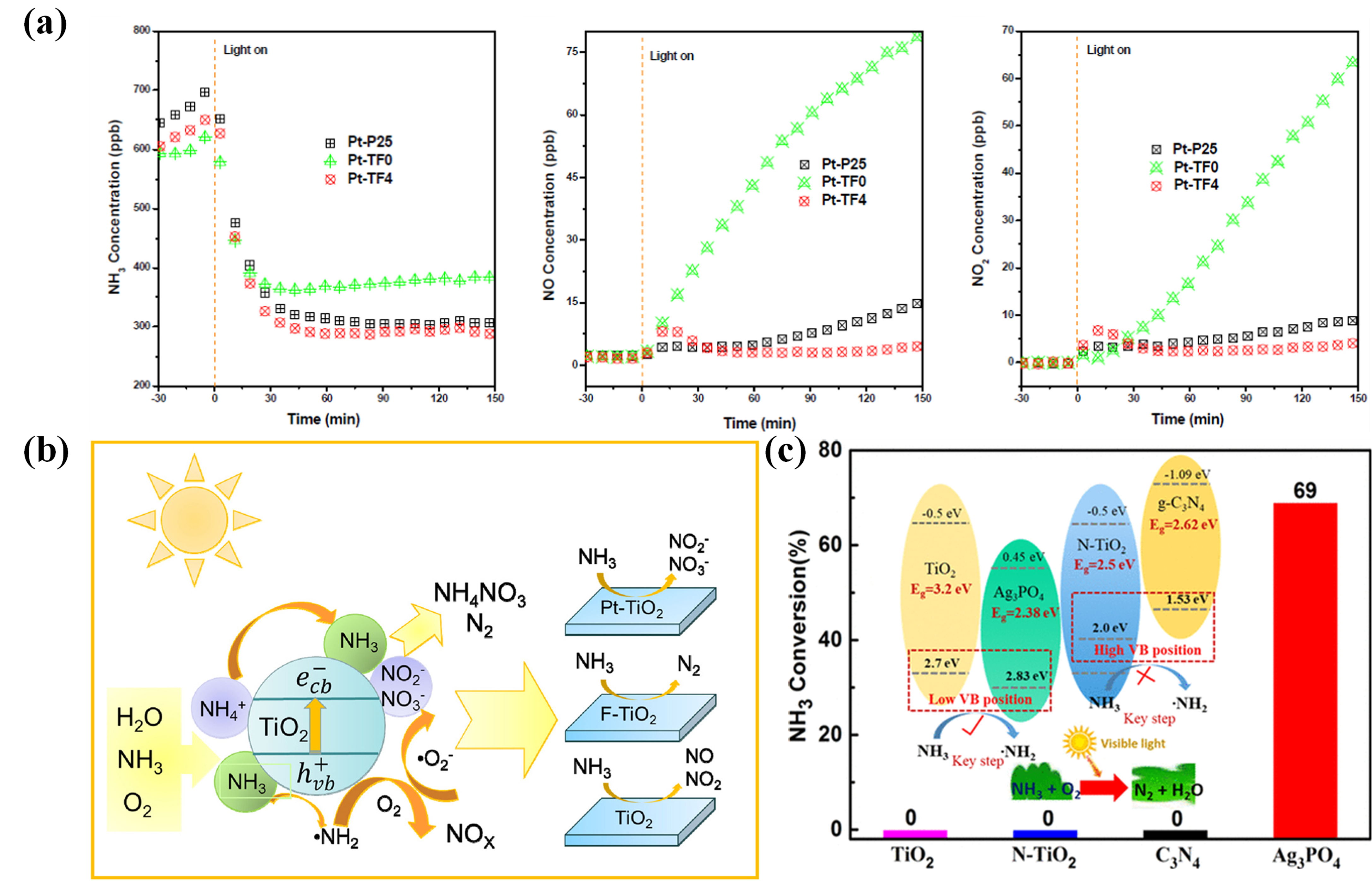
Shu et al. investigated the effect of F or Pt modification on the catalytic activity of TiO
2 (Pt/F−TiO
2) for selective photocatalytic oxidation of NH
3. They found that surface fluorination had no significant effect on the degradation efficiency of NH
3, but it could enhance the adsorption of NH
3 on the TiO
2 surface and might reduce the generation of harmful NO
x by influencing the reaction pathway (Figure 7a). In addition, the deposited Pt prolonged the lifetime of photogenerated carriers by strongly trapping electrons, and the electron-rich Pt acted as a reduction site, providing electrons to NO
x to promote N
2 production (Figure 7b) [
64].
Figure 7. (<strong>a</strong>) Outlet NH<sub>3</sub> concentration, NO concentration and NO<sub>2</sub> concentration of different samples (Pt/P25, Pt/TF0 and Pt/TF4) under simulated sunlight irradiation. (<strong>b</strong>) Schematic diagram of the photocatalytic oxidation process of NH<sub>3</sub> on Pt/F−TiO<sub>2</sub> [
64]. Reproduced with permission. Copyright 2022, Elsevier Publication. (<strong>c</strong>) Photocatalytic oxidation of NH<sub>3</sub> on different catalysts (TiO<sub>2</sub>, N−TiO<sub>2</sub>, C<sub>3</sub>N<sub>4</sub> and Ag<sub>3</sub>PO<sub>4</sub>) under visible light [
65]. Reproduced with permission. Copyright 2020, ACS Publication.
In addition to TiO
2 based catalysts, other photocatalysts have also been used for the photocatalytic oxidation of NH
3. He et al. prepared a series of typical visible light photocatalysts (N−TiO
2, g−C
3N
4 and Ag
3PO
4), hoping to realize the photocatalytic oxidation of NH
3 under visible light (Figure 7c). However, the results show that only Ag
3PO
4 has the removal effect in visible light. Therefore, an active, visible light photocatalyst for NH
3 oxidation requires both a suitable band gap for visible light response and a low VB edge associated with a high oxidation potential for activating NH
3 to ·NH
2. This result also confirmed that
h+ is responsible for triggering the initial key step of NH
3 oxidation [
65].
3. NH3 Removal Mechanism
At present, researchers have deeply studied the photocatalytic oxidation mechanism of NH
3, but only limited to TiO
2 based photocatalysts. First of all, adsorption is the first step in a chemical reaction. At present, researchers generally believe that the Lewis acidic site is the active site of NH
3 photocatalytic oxidation on TiO
2 based photocatalysts [
66]. NH
3 adsorbs at the Lewis acidic site will react with
h+ to form ·NH
2 for subsequent reaction under the irradiation of simulated sunlight (Equation (1)) [
67,
68].
Subsequently, three oxidation pathways are divided according to the different reactive substances with ·NH
2.
(i) ·NH
2 mechanism. ·NH
2 reacts with reactive oxygen species (ROS) to form NO
x or nitrate. This mechanism has been observed on almost all NH
3 photocatalytic oxidation catalysts. The researchers explored the photocatalytic oxidation path of NH
3 on anatase TiO
2 using in-situ DRIFTS. The results showed that the ·NH
2 reacted with the oxygen anion free radical (·O
2−) formed under ultraviolet irradiation to form NO. Further reacting with O
2, NO was converted to NO
2−, nitro and NO
3− (monodentate and bidentate) substances on the TiO
2 surface (Equations (2)–(7)) [
69].
(ii)
In-situ selective catalytic reduction (iSCR) mechanism. By observing the reaction products of ·NH
2 mechanism, it can be found that although NH
3 is removed by photocatalytic oxidation, NO
x will be generated, which is highly likely to cause secondary pollution to the environment. In addition, the formed nitrite and nitrate will cover the active site of the photocatalyst and lead to the deactivation of the catalyst. Fortunately, NH
3 can react with the formed NO
x and nitrate to form N
2 and water, the iSCR mechanism (Equations (8)–(10)). The existence of this mechanism promotes the improvement of nitrogen selectivity. Even if NO
x is generated in the process of NH
3 photocatalytic oxidation, the final products are N
2 and water [
70]. Yamazoe et al. found that ·NH
2 could selectively react with the formed monodentate and bidentate NO
3− and NO on TiO
2 to generate N
2 under UV light irradiation. The researchers also found a similar reaction path on Pt and F co-modified TiO
2 photocatalysts. Furthermore, it is proved that
h+ and
e− play an important role in the formation of NO
x through free radical quenching experiments. When ·OH and ·O
2− were removed, the production of NO
x increased significantly, indicating that ·OH and ·O
2− could inhibit the formation of NO
x or rapidly oxidize NO
x to nitrate and nitrite [
68]. Therefore, in the process of photocatalytic oxidation of NH
3, the above two mechanisms exist on almost all photocatalysts. That is, NH
3 is first oxidized to NO
x and nitrate by ·NH
2 mechanism and then reduced to harmless N
2 and water by the iSCR mechanism (Figure 8a,d,e).
(iii) N
2H
4 mechanism. The synergistic effect of these above two mechanisms seems to be able to treat NH
3 into harmless N
2 and water, but due to the mismatch in the rates of the two reactions, the generated NO
x cannot be reduced to N
2 in time, and part of NO
x and nitrate will still be produced, resulting in secondary pollution. In addition, it remains to be discussed whether nitrate can be reduced to N
2 (Equation (10)), so that nitrate substances remaining on the surface will cover the active site of the reaction and lead to the deactivation of the catalyst. In this case, a new mechanism of NH
3 oxidation was discovered and proposed by the researchers. That is, ·NH
2 is coupled to form N
2H
4 species (Figure 8d), followed by dehydrogenation to form N
2H
2 species, and finally dehydrogenation to form N
2 and water (Equations (11)–(13)). The N
2H
4 pathway, which neither involves the formation of NO
x and nitrates nor leads to secondary environmental pollution and catalyst deactivation, appears to be an economically and environmentally friendly route for NH
3 removal. In the exploration of selective catalytic oxidation of NH
3, the N
2H
4 mechanism was first discovered on the Al
2O
3 catalyst supported by Pd or Ag in thermal catalytic oxidation [
28,
30]. Using in-situ characterization techniques, the researchers found a characteristic peak at 2170 cm
−1 that gradually increased with the response and attributed it to the HN=NH species. Recently, the N
2H
4 mechanism has also been observed during the photocatalytic oxidation of NH
3 on Mo, C−TiO
2 photocatalyst, and it has been confirmed that the introduction of Mo ions is responsible for the existence of this mechanism [
52]. Additionally, in the investigation of electrocatalytic oxidation of NH
3, the N
2H
4 mechanism has also been observed on Ru-based catalysts and the high value-added substance N
2H
4 is used as the end product (Figure 9b) [
71]. In fact, the researchers also found the N
2H
4 mechanism in the reaction process of photocatalytic cracking of NH
3. The decomposition mechanism of NH
3 on Ni/TiO
2 under UV irradiation was calculated using DFT simulation (Figure 9a) [
72]. The researchers simulated three NH
3 decomposition paths and calculated the activation energy of each step separately. The results showed that the maximum activation energy of route 1 is 235.7 KJ/mol, which made it almost impossible for Route 1 to react. In contrast, the highest activation energy of route 2 and route 2′ is 74.4 and 59.2 KJ/mol, respectively. Therefore, the decomposition path of NH
3 should be first coupled to generate N
2H
4 and then gradually dehydrogenation.
Generally, researchers have identified three mechanisms for NH
3 removal during photocatalytic oxidation: the ·NH
2 mechanism, the iSCR mechanism and the N
2H
4 mechanism. Notably, the ·NH
2 and the iSCR mechanisms often coexist in most photocatalytic oxidation processes for ammonia. In the ·NH
2 mechanism, the ·NH
2 species is oxidized to NO, NO
2 or nitrate. Subsequently, the generated NO
x reacts with ·NH
2 molecules via the iSCR mechanism, resulting in the production of harmless N
2 and water, thereby ensuring N
2 selectivity in the photocatalytic process. However, the mismatch between the reaction rates of these two mechanisms can easily lead to the release of NO
x and the deactivation of the catalyst due to nitrate accumulation on the catalyst surface. In response to this challenge, the N
2H
4 mechanism has emerged as a promising alternative for ammonia removal. The N
2H
4 species can gradually dehydrogenate to produce N
2 and water without generating NO
x or nitrate, thereby preventing secondary environmental pollution and reducing catalyst deactivation.
In addition, humidity also has an important effect on the NH
3 oxidation mechanism under environmental conditions. The researchers explored the path of photocatalytic oxidation of ppb level NH
3 on TiO
2 and explored the role of water in the process. Below 50% relative humidity, water catalyzed reactions that promote NO
x formation. However, above 50%, the increase in adsorbed water hindered the contact of active sites, promoted the formation of non-reactive NH
4+, and reduced oxidant levels, thereby decreasing NO
x formation [
73]. The researchers used a coated wall flow tube and a chemiluminescence NO
x analyzer to study the kinetics of NH
3 absorption and NO
x formation on irradiated TiO
2 surfaces. They found that NH
3 absorption kinetics are inversely proportional to NH
3 concentration, indicating adherence to the Langmuir-Hinshelwood mechanism. The first step of the reaction involved a collision reaction between NH
3 and valence band holes on the TiO
2 surface, generating ·NH
2. ·NH
2 reacted with O
2 to form amino peroxy radicals (NH
2OO), a reaction that proceeded more rapidly in the aqueous phase. NH
2OO undergoes water-catalyzed isomerization and decomposition reactions to produce NO, which was further oxidized to NO
2 on the TiO
2 surface. Water plays a crucial catalytic role throughout the reaction mechanism (Figure 8c). Theoretical calculations supported the experimental results, indicating that the solvation of key intermediates facilitates proton transfer isomerization, making the NO formation step exothermic. The NO
x produced by the P25 photocatalyst was twice that of anatase, while the reactivity of rutile was less than 10% of anatase. This is due to differences in their E
g, adsorption capacity, and electron-hole recombination efficiency.
Figure 8. (<strong>a</strong>) Mechanism of photocatalytic oxidation of NH<sub>3</sub> on TiO<sub>2</sub> [
69]. Reproduced with permission. Copyright 2008, Elsevier Publication. (<strong>b</strong>) NH<sub>3</sub> photooxidation in air forms HONO on TiO<sub>2</sub> when RH = 48% [
74]. Reproduced with permission. (<strong>c</strong>) NH<sub>3</sub> will be photo-oxidized to NO<sub>x</sub> on TiO<sub>2</sub> under the catalysis of water [
73]. Copyright 2013, ACS Publication. (<strong>d</strong>) The proposed N<sub>2</sub>H<sub>4</sub> mechanism of NH<sub>3</sub> photo-oxidation on Mo, C−TiO<sub>2</sub> [
52]. (<strong>e</strong>) Mechanism of photocatalytic oxidation of NH<sub>3</sub> on Pt/F−TiO<sub>2</sub> (·NH<sub>2</sub> and iSCR mechanism) [
64]. Reproduced with permission. Copyright 2022 and 2024, Elsevier Publication.
In other work, Kebede et al. indicated that (Figure 8b) TiO
2 could convert NH
3 into HONO under light exposure, which resulted from the reduction of NO
2 and NO
3− produced by the photooxidation of NH
3 catalyzed by water. HONO is a form of NO
x in the atmosphere that can generate ozone and other secondary pollutants through photochemical reactions, thereby exacerbating air pollution [
74]. Exposure to high concentrations of HONO may have adverse effects on human health. It can irritate the respiratory tract and trigger or worsen asthma and other respiratory diseases. The results showed that the amount of HONO formed depends on the initial NH
3 concentration and the relative humidity of the carrier gas. The highest HONO yield, reaching 350 ppb, was observed at moderate NH
3 concentrations (150–290 ppb) and 30–40% relative humidity. However, when the relative humidity increased from 40% to 95%, the HONO yield decreased from 350 ppt to 50 ppt. This was consistent with previous studies on the photocatalytic oxidation of NH
3 to nitrogen dioxide by TiO
2, suggesting that at relative humidity below 40%, water-promoted reactions favor the formation of nitrogen dioxide. In contrast, at relative humidity above 40%, capillary condensation filled the micropores on the TiO
2 surface, hindering access to reactive sites and thus reducing the formation of NO
2 and HONO. When NH
3 concentration exceeded 290 ppb, the formation of HONO decreased, possibly due to the reaction (iSCR mechanism) between HONO and NH
3.
Figure 9. (<strong>a</strong>) Reaction mechanism for NH<sub>3</sub> decomposition to N<sub>2</sub> and H<sub>2</sub> over TiO<sub>2</sub> photocatalyst [
72]. Reproduced with permission. Copyright 2017, Elsevier Publication. (<strong>b</strong>) Mechanism of electrocatalytic oxidation of NH<sub>3</sub> to N<sub>2</sub>H<sub>4</sub> [
71]. Reproduced with permission. Copyright 2023, Springer Nature Publication. (<strong>c</strong>) Mechanism of photothermal synergistic catalytic oxidation of NH<sub>3</sub> and oxidation path of NH<sub>3</sub> [
75]. Reproduced with permission. Copyright 2023, Elsevier Publication.
Therefore, optimal relative humidity (RH) levels can enhance the removal efficiency of photocatalyzed NH
3 oxidation. However, excessive RH can hinder the adsorption of NH
3 molecules due to the presence of water, which simultaneously catalyzes the formation of NO
x. This interaction can lead to a reduction in both the reaction activity of the catalyst and its selectivity for N
2.
4. Challenges and Prospects of Photocatalytic Oxidation of NH3
Photocatalytic oxidation as a promising NH
3 treatment strategy is being gradually improved, but there are still many challenges. However, the catalysts for the photocatalytic oxidation of NH
3 have been studied in depth, including the performance and oxidation mechanism. However, it is only limited to TiO
2 based catalysts, and other kinds of photocatalysts are rarely studied. In addition, due to the narrow range of photo-response and low utilization of charge carriers, the removal efficiency of photocatalytic oxidation does not meet the requirements of practical applications. Furthermore, NO
x or nitrate produced by the ·NH
2 mechanism cannot be reduced to N
2 and water in time, resulting in insufficient nitrogen selectivity of the reaction products. As a result, the accumulated NO
x may cause secondary pollution to the air, and the nitrate remaining on the surface of the photocatalyst will lead to the deactivation of the catalyst, increasing the cost of treatment. To solve the above problems, researchers use the heat generated by light irradiation to improve the utilization rate of solar energy and charge carriers through photothermal synergistic catalysis. For a flow with an NH
3 concentration of about 45 ppm, the NH
3 conversion rate of 91.7% and N
2 selectivity of 94.7% were obtained on the Cryptomelane nanowires (Figure 9c) [
75]. Since photothermal co-catalysis had higher solar energy and carrier utilization than photocatalysis alone, it seems to be more suitable for ammonia treatment. However, it could be seen from the experimental results of cyclic on-off light that the photocatalyst still had an obvious deactivation phenomenon. This was because the experimental results confirmed that the oxidation mechanism of NH
3 was the coexistence of ·NH
2 and iSCR mechanism, indicating that there was still the formation of NO
x and nitrate, which may still cause secondary environmental pollution and catalyst deactivation. Therefore, the development of photocatalysts with N
2H
4 mechanism as the main oxidation path seems to be the key to solving this problem. Because NH
3 oxidation removal through this mechanism will not produce NO
x resulting in secondary pollution of the environment, nor will nitrate occupy the reactive active site.
In summary, with the gradual increase in human demand for NH
3, the environmental pollution caused by NH
3 is becoming increasingly serious. Due to the characteristics of discrete emission and low concentration, the traditional methods of treating gaseous pollutants are difficult to remove and high cost. In contrast, photocatalytic oxidation is an economical and practical strategy. However, the existing photocatalyst solar energy and carrier utilization rate are not high enough, resulting in the removal effect of NH
3 is not ideal. Currently, traditional methods such as metal loading, semiconductor compounding, and surface modification are employed to enhance ammonia removal efficiency and N
2 selectivity. Additionally, further research is needed to explore the application of other emerging photocatalyst modification strategies in the photocatalytic oxidation of NH
3, including semiconductor quantum dots, alloy loading, and metal phosphides [
76,
77]. In addition, the ·NH
2 mechanism will produce NO
x and nitrate. Although the iSCR mechanism will reduce part of NO
x to produce N
2 and water, it may still cause secondary environmental pollution and catalyst deactivation. Therefore, the exploration of photocatalysts with high removal performance and N
2H
4 mechanism as the NH
3 oxidation path may be a promising strategy to solve the NH
3 pollution problem in the future.
Acknowledgments
The authors express gratitude to the National Key Technology Research and Development Project of China (2019YFC1906404) for providing financial support for this study.
Author Contributions
Y.W.: Writing—original draft, Investigation, Formal analysis. Y.Y.: Writing—review & editing. Y.G.: Writing—review & editing, Investigation. S.Y.: Writing—review & editing, Supervision, Resources, Project administration, Funding acquisition, Conceptualization.
Ethics Statement
Not applicable.
Informed Consent Statement
Not applicable.
Funding
This research was funded by the National Key Technology Research and Development Project of China (2019YFC1906404).
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
1.
Shu Y, Wang D, Wang J, Huang H. Adsorption and photocatalytic degradation of Ammonia: Status and challenges.
Chem. Eng. J. 2024,
498, 154925.
[Google Scholar]
2.
Afif A, Radenahmad N, Cheok Q, Shams S, Kim JH, Azad AK. Ammonia−fed fuel cells: a comprehensive review. Renew.
Sustain. Energy Rev. 2016,
60, 822–835.
[Google Scholar]
3.
Berwal P, Kumar S, Khandelwal B. A comprehensive review on synthesis, chemical kinetics, and practical application of ammonia as future fuel for combustion.
J. Energy Inst. 2021,
99, 273–298.
[Google Scholar]
4.
Jeerh G, Zhang M, Tao S. Recent progress in ammonia fuel cells and their potential applications.
J. Mater. Chem. A 2021,
9, 727–752.
[Google Scholar]
5.
Jiang J, Gao F, Wang S, Tang X, Lu M, Wang J, et al. Advances in photo-catalytic oxidation of NH
3 over modified TiO
2 catalysts: Reaction pathways, improvement strategy and promotion mechanism.
J. Environ. Chem. Eng. 2023,
11, 110602.
[Google Scholar]
6.
Vikrant K, Kim K−H, Dong F, Giannakoudakis DA. Photocatalytic Platforms for Removal of Ammonia from Gaseous and Aqueous Matrixes: Status and Challenges.
ACS Catal. 2020,
10, 8683–8716.
[Google Scholar]
7.
Xu P, Li G, Zheng Y, Fung JCH, Chen A, Zeng Z, et al. Fertilizer management for global ammonia emission reduction.
Nature 2024,
626, 792–798.
[Google Scholar]
8.
Chang Y, Zou Z, Zhang Y, Deng C, Hu J, Shi Z, et al. Assessing Contributions of Agricultural and Nonagricultural Emissions to Atmospheric Ammonia in a Chinese Megacity.
Environ. Sci. Technol. 2019,
53, 1822–1833.
[Google Scholar]
9.
Nie E, Zheng G, Shao Z, Yang J, Chen T. Emission characteristics and health risk assessment of volatile organic compounds produced during municipal solid waste composting.
Waste Manag. 2018,
79, 188–195.
[Google Scholar]
10.
Vikrant K, Roy K, Kim K−H, Bhattacharya SS. Insights into the storage stability of ammonia in polyester aluminum bags.
Environ. Res. 2019,
177, 108596.
[Google Scholar]
11.
Schwartz−Narbonne H, Jones SH, Donaldson DJ. Indoor Lighting Releases Gas Phase Nitrogen Oxides from Indoor Painted Surfaces.
Environ. Sci. Technol. Lett. 2019,
6, 92–97.
[Google Scholar]
12.
Photiou P, Kallis M, Samanides CG, Vyrides I, Padoan E, Montoneri E, et al. Integrated Chemical Biochemical Technology to Reduce Ammonia Emission from Fermented Municipal Biowaste.
Environ. Sci. Technol. Lett. 2021,
9, 8402–8413.
[Google Scholar]
13.
Liu T, Wang X, Wang B, Ding X, Deng W, Lü S, et al. Emission factor of ammonia (NH
3) from on−road vehicles in China: Tunnel tests in urban Guangzhou.
Environ. Res. Lett. 2014,
9, 064027.
[Google Scholar]
14.
Huang C, Hu Q, Lou S, Tian J, Wang R, Xu C, et al. Ammonia Emission Measurements for Light−Duty Gasoline Vehicles in China and Implications for Emission Modeling.
Environ. Res. Lett. 2018,
52, 11223–11231.
[Google Scholar]
15.
Hopke PK, Querol X. Is Improved Vehicular NO
x Control Leading to Increased Urban NH
3 Emissions?
Environ. Res. Lett. 2022,
56, 11926–11927.
[Google Scholar]
16.
Farren NJ, Davison J, Rose RA, Wagner RL, Carslaw DC. Underestimated Ammonia Emissions from Road Vehicles.
Envi-ron. Res. Lett. 2020,
54, 15689–15697.
[Google Scholar]
17.
Chen Z−L, Song W, Hu C−C, Liu X−J, Chen G−Y, Walters WW, et al. Significant contributions of combustion−related sources to ammonia emissions.
Nat. Commun. 2022,
13, 7710.
[Google Scholar]
18.
Xu W, Zhao Y, Wen Z, Chang Y, Pan Y, Sun Y, et al. Increasing importance of ammonia emission abatement in PM2.5 pollution control.
Sci. Bull. 2022,
67, 1745–1749.
[Google Scholar]
19.
Liu Y, Zhan J, Zheng F, Song B, Zhang Y, Ma W, et al. Dust emission reduction enhanced gas−to−particle conversion of ammonia in the North China Plain.
Nat. Commun. 2022,
13, 6887.
[Google Scholar]
20.
Gu B, Zhang L, Van Dingenen R, Vieno M, Van Grinsven HJ, Zhang X, et al. Abating ammonia is more cost−effective than nitrogen oxides for mitigating PM2.5 air pollution.
Science 2021,
374, 758–762.
[Google Scholar]
21.
Liu Z, Rieder HE, Schmidt C, Mayer M, Guo Y, Winiwarter W, et al. Optimal reactive nitrogen control pathways identified for cost−effective PM2.5 mitigation in Europe.
Nat. Commun. 2023,
14, 4246.
[Google Scholar]
22.
Zhou S, Li Y, Liao X, Wang W, Mao C, Mi J, et al. A low−cost deodorizing spray net device for the removal of ammonia emissions in livestock houses.
J. Clean. Prod. 2021,
318, 128516.
[Google Scholar]
23.
Hu T−T, Liu F, Dou S, Zhong L−B, Cheng X, Shao Z−D, et al. Selective adsorption of trace gaseous ammonia from air by a sulfonic acid−modified silica xerogel: Preparation, characterization and performance.
Chem. Eng. J. 2022,
443, 136357.
[Google Scholar]
24.
Gebreegziabher TB, Wang S, Nam H. Adsorption of H
2S, NH
3 and TMA from indoor air using porous corncob activated carbon: Isotherm and kinetics study.
J. Environ. Chem. Eng. 2019,
7, 103234.
[Google Scholar]
25.
Han X, Lu W, Chen Y, da Silva I, Li J, Lin L, et al. High Ammonia Adsorption in MFM−300 Materials: Dynamics and Charge Transfer in Host–Guest Binding.
J. Am. Chem. Soc. 2021,
143, 3153–3161.
[Google Scholar]
26.
Ma B, LaPara TM, Kim T, Hozalski RM. Multi−scale Investigation of Ammonia−Oxidizing Microorganisms in Biofilters Used for Drinking Water Treatment.
J. Am. Chem. Soc. 2023,
57, 3833–3842.
[Google Scholar]
27.
Liu J, Li X, Xu Y, Wu Y, Wang R, Zhang X, et al. Highly efficient reduction of ammonia emissions from livestock waste by the synergy of novel manure acidification and inhibition of ureolytic bacteria.
Environ. Int. 2023,
172, 107768.
[Google Scholar]
28.
Wang F, Ma J, He G, Chen M, Zhang C, He H. Nanosize Effect of Al
2O
3 in Ag/Al2O3 Catalyst for the Selective Catalytic Oxidation of Ammonia.
ACS Catal. 2018,
8, 2670–2682.
[Google Scholar]
29.
Wang F, He G, Zhang B, Chen M, Chen X, Zhang C, et al. Insights into the Activation Effect of H
2 Pretreatment on Ag/Al
2O
3 Catalyst for the Selective Oxidation of Ammonia.
ACS Catal. 2019,
9, 1437–1445.
[Google Scholar]
30.
Dann EK, Gibson EK, Blackmore RH, Catlow CRA, Collier P, Chutia A, et al. Structural selectivity of supported Pd nano-particles for catalytic NH
3 oxidation resolved using combined operando spectroscopy.
Nat. Catal. 2019,
2, 157–163.
[Google Scholar]
31.
Kobayashi H, Hayakawa A, Somarathne KDKA, Okafor EC. Science and technology of ammonia combustion.
Proc. Combust. Inst. 2019,
37, 109–133.
[Google Scholar]
32.
Kuk SK, Ji SM, Kang S, Yang DS, Kwon HJ, Koo MS, et al. Singlet−oxygen−driven photocatalytic degradation of gaseous formaldehyde and its mechanistic study.
Appl. Catal. B 2023,
328, 122463.
[Google Scholar]
33.
Fan H, Wang R. Low−temperature NH
3−SCR reaction over 3D Cu/Fe−TiO
2−rGO composite catalyst synthesized by photo-reduction method.
Appl. Catal. B 2022,
450, 138152.
[Google Scholar]
34.
Guo Q, Zhou C, Ma Z, Ren Z, Fan H, Yang X. Elementary photocatalytic chemistry on TiO
2 surfaces.
Chem. Soc. Rev. 2016,
45, 3701–3730.
[Google Scholar]
35.
Akhter P, Nawaz S, Shafiq I, Nazir A, Shafique S, Jamil F, et al. Efficient visible light assisted photocatalysis using ZnO/TiO
2 nanocomposites.
Mol. Catal. 2023,
535, 112896.
[Google Scholar]
36.
Guo L, Zhang J, Zhang X, Wang R, Jia Y, Long H. Energy band matching Bi
2WO
6/black−TiO
2 Z−scheme heterostructure for the enhanced visible−light photocatalytic degradation of toluene.
Mol. Catal. 2023,
550, 113603.
[Google Scholar]
37.
Shang F−K, Li Y−H, Qi M−Y, Tang Z−R, Xu Y−J. Photocatalytic materials for sustainable chemistry via cooperative pho-toredox catalysis.
Catal. Today 2023,
410, 85–101.
[Google Scholar]
38.
Zhang Y, Qi M−Y, Tang Z−R, Xu Y−J. Photoredox−Catalyzed Plastic Waste Conversion: Nonselective Degradation versus Selective Synthesis.
ACS Catal. 2023,
13, 3575–3590.
[Google Scholar]
39.
Huang L, He G, Yuan Y, Zhang TC, Wang Y, Yuan S. Trivalent Metal Ions (Al, Ga, In)−Doped TiO
2 for Enhanced Photo-catalytic Desulfurization of H
2S: Band Structure Regulation, Performance, and Mechanism.
Ind. Eng. Chem. Res. 2024,
63, 7154–7165.
[Google Scholar]
40.
Huang L, Yuan Y, Wang Y, Yılmaz M, Zhang TC, Yuan S. Visible−Light−Driven photocatalytic oxidation of H
2S by Bo-ron−doped TiO
2/LDH Heterojunction: Synthesis, performance, and reaction mechanism.
Chem. Eng. J. 2022,
448, 137607.
[Google Scholar]
41.
Guo Q, Zhou C, Ma Z, Yang X. Fundamentals of TiO
2 Photocatalysis: Concepts, Mechanisms, and Challenges.
Adv. Mater. 2019,
31, 1901997.
[Google Scholar]
42.
Wu H, Ma J, Zhang C, He H. Effect of TiO
2 calcination temperature on the photocatalytic oxidation of gaseous NH
3.
J. Environ. Sci. 2014,
26, 673–682.
[Google Scholar]
43.
Heylen S, Smet S, Laurier KGM, Hofkens J, Roeffaers MBJ, Martens JA. Selective photocatalytic oxidation of gaseous ammonia to dinitrogen in a continuous flow reactor.
Catal. Sci. Technol. 2012,
2, 1802.
[Google Scholar]
44.
Sola AC, Sousa DG, Araña J, Díaz OG, Rodríguez JMD, de la Piscina PR, et al. Differences in the vapour phase photocata-lytic degradation of ammonia and ethanol in the presence of water as a function of TiO
2 characteristics and the presence of O
2.
Catal. Today 2016,
266, 53–61.
[Google Scholar]
45.
Wu H, Ma J, Li Y, Zhang C, He H. Photocatalytic oxidation of gaseous ammonia over fluorinated TiO
2 with exposed (001) facets.
Appl. Catal. B 2014,
152–153, 82–87.
[Google Scholar]
46.
Chen M, Ma J, Zhang B, He G, Li Y, Zhang C, et al. Remarkable synergistic effect between {001} facets and surface F ions promoting hole migration on anatase TiO
2.
Appl. Catal. B 2017,
207, 397–403.
[Google Scholar]
47.
Chen M, Ma J, Zhang B, Wang F, Li Y, Zhang C, et al. Facet−dependent performance of anatase TiO
2 for photocatalytic oxidation of gaseous ammonia.
Appl. Catal. B 2018,
223, 209–215.
[Google Scholar]
48.
Saoud WA, Assadi AA, Guiza M, Bouzaza A, Aboussaoud W, Soutrel I, et al. Abatement of ammonia and butyraldehyde under non−thermal plasma and photocatalysis: Oxidation processes for the removal of mixture pollutants at pilot scale.
Chem. Eng. J. 2018,
344, 165–172.
[Google Scholar]
49.
Wang P, Shen Z, Xia Y, Wang H, Zheng L, Xi W, et al. Atomic Insights for Optimum and Excess Doping in Photocatalysis: A Case Study of Few−Layer Cu−ZnIn
2S
4.
Adv. Funct. Mater. 2019,
29, 1807013.
[Google Scholar]
50.
Wang S, Yu H, Cheng X. Degradation of Typical Indoor Air Pollutants Using Fe−Doped TiO
2 Thin Film under Daylight Illu-mination.
J. Chem. 2014,
2014, 1–5.
[Google Scholar]
51.
Sirivallop A, Areerob T, Chiarakorn S. Enhanced Visible Light Photocatalytic Activity of N and Ag Doped and Co−Doped TiO2 Synthesized by Using an In−Situ Solvothermal Method for Gas Phase Ammonia Removal.
Catalysts 2020,
10, 251.
[Google Scholar]
52.
Wang Y, Huang L, Zhang TC, Wang Y, Yuan S. Visible−Light−Induced photocatalytic oxidation of gaseous ammonia on Mo, c−codoped TiO
2: Synthesis, performance and mechanism.
Chem. Eng. J. 2024,
482, 148811.
[Google Scholar]
53.
Jiang J, Gao F, Zhang J, Lu M, Sun L, Lei Y, et al. Enhancing activity and non−deactivating stability on N−modified TiO2 catalyst for visible−light photocatalytic oxidation of ammonia at room temperature.
Appl. Surf. Sci. 2024,
651, 159238.
[Google Scholar]
54.
Gao F, Zhang J, Jiang J, Tang X, Zhou Y, Yi H. Visible light−induced photocatalytic oxidation of gaseous ammonia on C surface−coated N−TiO
2 catalyst: Synthesis, properties and mechanism.
Appl. Surf. Sci. 2025,
358, 130349.
[Google Scholar]
55.
Pu S, Wang H, Zhu J, Li L, Long D, Jian Y, et al. Heterostructure Cu
2O/(001)TiO
2 Effected on Photocatalytic Degradation of Ammonia of Livestock Houses.
Catalysts 2019,
9, 267.
[Google Scholar]
56.
Zhu J, Jian Y, Long D, Wang H, Zeng Y, Li J, et al. Degradation of ammonia gas by Cu
2O/{001}TiO
2 and its mechanistic analysis.
RSC Adv. 2021,
11, 3695–3702.
[Google Scholar]
57.
Čižmar T, Grčić I, Bohač M, Razum M, Pavić L, Gajović A. Dual Use of Copper−Modified TiO
2 Nanotube Arrays as Mate-rial for Photocatalytic NH
3 Degradation and Relative Humidity Sensing.
Coatings 2021,
11, 1500.
[Google Scholar]
58.
Chen M, Chen J, Chen C, Zhang C, He H. Distinct photocatalytic charges separation pathway on CuO
x modified rutile and anatase TiO
2 under visible light.
Appl. Catal. B 2022,
300, 120735.
[Google Scholar]
59.
Pu S, Long D, Liu Z, Yang F, Zhu J. Preparation of RGO−P25 Nanocomposites for the Photocatalytic Degradation of Am-monia in Livestock Farms.
Catalysts 2018,
8, 189.
[Google Scholar]
60.
Gao F, Song S, Tang X, Yi H, Zhao S, Yu Q. Tetraphenyl–porphyrin decorated anatase TiO
2 catalysts for the visible–light photocatalytic oxidation of gaseous ammonia at room temperature.
Appl. Surf. Sci. 2020,
506, 144421.
[Google Scholar]
61.
Zhang H, Gu Q−Q, Zhou Y−W, Liu S−Q, Liu W−X, Luo L, et al. Direct Z−scheme photocatalytic removal of ammonia via the narrow band gap MoS
2/N−doped graphene hybrid catalyst upon near−infrared irradiation.
Appl. Surf. Sci. 2020,
504, 144065.
[Google Scholar]
62.
Li Z, Li D, Feng Z, Lv S, Zhang Q, Yu Y, et al. Enhanced photocatalytic ammonia oxidation over WO3@TiO2 heterostruc-tures by constructing an interfacial electric field.
Chemosphere 2024,
355, 141811.
[Google Scholar]
63.
Li Y−N, Chen Z−Y, Bao S−J, Wang M−Q, Song C−L, Pu S, et al. Ultrafine TiO
2 encapsulated in nitrogen−doped porous carbon framework for photocatalytic degradation of ammonia gas.
Chem. Eng. J. 2018,
331, 383–388.
[Google Scholar]
64.
Shu Y, Ji J, Zhou M, Liang S, Xie Q, Li S, et al. Selective photocatalytic oxidation of gaseous ammonia at ppb level over Pt and F modified TiO
2.
Appl. Catal. B 2022,
300, 120688.
[Google Scholar]
65.
Chen M, Zhang C, He H. Insights into Designing Photocatalysts for Gaseous Ammonia Oxidation under Visible Light.
Environ. Sci. Technol. 2020,
54, 10544–10550.
[Google Scholar]
66.
Bühlmeyer H, Adamsen KC, Xu T, Lammich L, Libuda J, Lauritsen JV, et al. Adsorption and Reaction of NH
3 on Rutile TiO
2 (110): An STM Study.
J. Phys. Chem. C 2022,
126, 6590–6600.
[Google Scholar]
67.
Yamazoe S, Teramura K, Hitomi Y, Shishido T, Tanaka T. Visible Light Absorbed NH
2 Species Derived from NH
3 Adsorbed on TiO
2 for Photoassisted Selective Catalytic Reduction.
J. Phys. Chem. C 2007,
111, 14189–14197.
[Google Scholar]
68.
Yamazoe S, Okumura T, Hitomi Y, Shishido T, Tanaka T. Mechanism of Photo−Oxidation of NH
3 over TiO
2: Fourier Transform Infrared Study of the Intermediate Species.
J. Phys. Chem. C 2007,
111, 11077–11085.
[Google Scholar]
69.
Yamazoe S, Hitomi Y, Shishido T, Tanaka T. Kinetic study of photo−oxidation of NH
3 over TiO
2.
Appl. Catal. B 2008,
82, 67–76.
[Google Scholar]
70.
Kolinko PA, Kozlov DV. Products distribution during the gas phase photocatalytic oxidation of ammonia over the various titania based photocatalysts.
Appl. Catal. B 2009,
90, 126–131.
[Google Scholar]
71.
Chen G, He P, Liu C, Mo X−F, Wei J−J, Chen Z−W, et al. Direct synthesis of hydrazine by efficient electrochemical ruthenium−catalysed ammonia oxidation.
Nat. Catal. 2023,
6, 949–958.
[Google Scholar]
72.
Utsunomiya A, Okemoto A, Nishino Y, Kitagawa K, Kobayashi H, Taniya K, et al. Mechanistic study of reaction mechanism on ammonia photodecomposition over Ni/TiO
2 photocatalysts.
Appl. Catal. B 2017,
206, 378–383.
[Google Scholar]
73.
Kebede MA, Varner ME, Scharko NK, Gerber RB, Raff JD. Photooxidation of Ammonia on TiO
2 as a Source of NO and NO
2 under Atmospheric Conditions.
J. Am. Chem. Soc. 2013,
135, 8606–8615.
[Google Scholar]
74.
Kebede MA, Scharko NK, Appelt LE, Raff JD. Formation of Nitrous Acid during Ammonia Photooxidation on TiO
2 under Atmospherically Relevant Conditions.
J. Am. Chem. Soc. 2013,
4, 2618–2623.
[Google Scholar]
75.
Zhou Y, Feng Y, Xie H, Lu J, Ding D, Rong S. Cryptomelane nanowires for highly selective self−heating photothermal synergistic catalytic oxidation of gaseous ammonia.
Appl. Catal. B 2023,
331, 122668.
[Google Scholar]
76.
Wu H-L, Qi M-Y, Tang Z-R, Xu Y-J. Semiconductor quantum dots: A versatile platform for photoredox organic transformation.
J. Mater. Chem. A 2023,
11, 3262–3280.
[Google Scholar]
77.
Li S-H, Qi M-Y, Tang Z-R, Xu Y-J. Nanostructured metal phosphides: From controllable synthesis to sustainable catalysis.
Chem. Soc. Rev. 2021,
50, 7539–7586.
[Google Scholar]