1.1. TGF-β1 Activation and Signaling
One of the most prominent inducers of fibrosis is the inflammatory cytokine Transforming Growth Factor-β1 (TGF-β1). In addition to its role as a modulator of immune system function (reviewed in [
1]), TGF-β1 has been shown to play a significant role in a host of pathologies, including cancer tumorigenesis and metastasis, chronic kidney disease, idiopathic lung disease, cardiac disease, osteogenesis imperfecta, and Parkinson’s disease [
2,
3,
4,
5,
6,
7]. It is expressed in almost all tissues in the body (reviewed in [
8]), and its expression is regulated by numerous signaling pathways (reviewed in [
9]).
TGF-β1 and its two related isoforms, TGF-β2 and TGF-β3, are secreted as latent complexes comprised of 3 components: a mature TGF-β ligand non-covalently bound to a latency-associated peptide (LAP), which is then bound to a latent TGF-β binding protein (LTBP). LTBP is able to bind to the extracellular matrix (ECM) protein fibronectin (FN) through cryptic growth factor binding sites on tensed FN, acting as a reservoir for latent TGF-β1 [
10,
11,
12,
13,
14,
15,
16,
17]. Active TGF-β1 can be released from the latent complex via decreases in pH, proteolysis by extracellular enzymes such as matrix metalloproteinases, or traction force-mediated opening of the LAP through αv integrins, most notably integrins αvβ1/3/6/8 [
12,
17,
18,
19]. These integrins are often clustered into large enzymatic complexes known as focal adhesions that coordinate extracellular attachments with the intracellular cytoskeleton (). It is through these focal adhesions that intracellular, acto-myosin based traction forces are transduced to manipulate the ECM, regulate cell size, and coordinate cellular motility [
12,
14,
20,
21,
22].
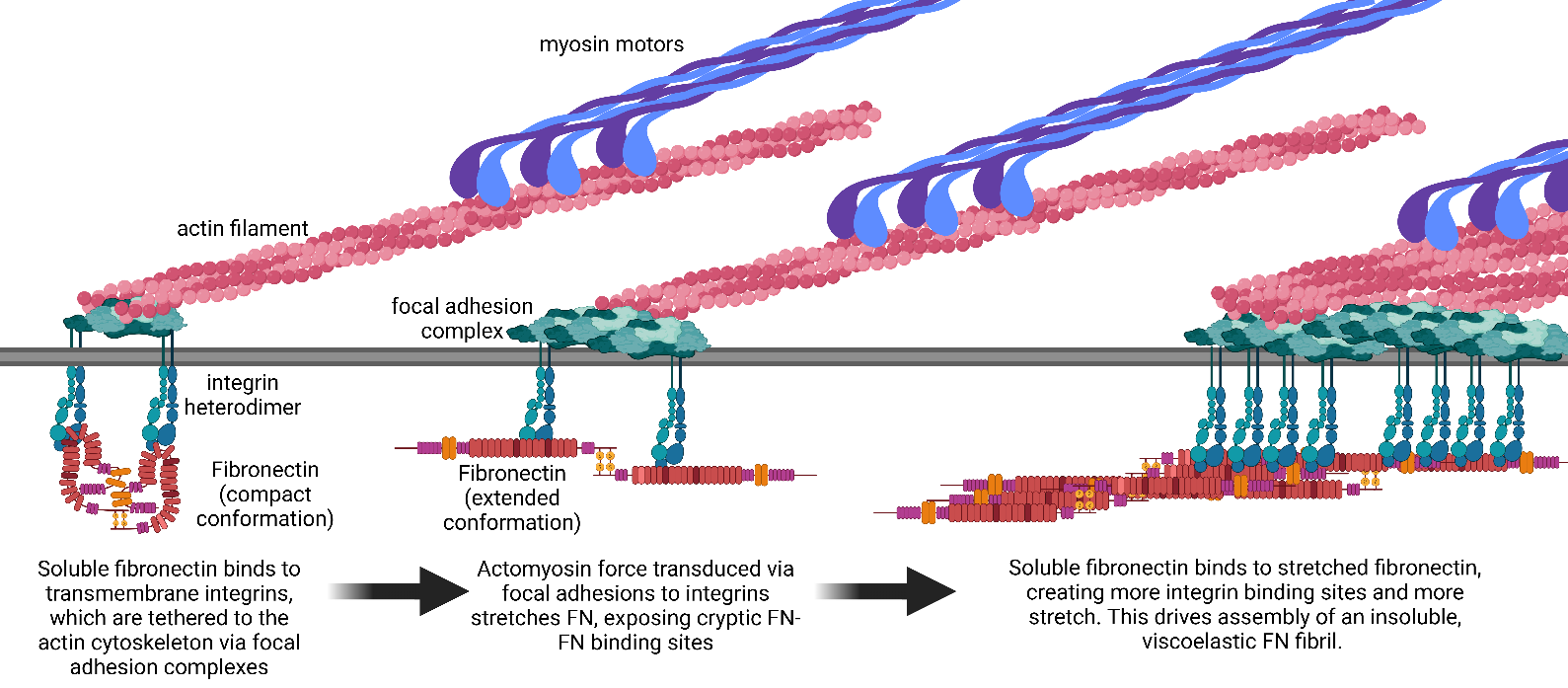
. Assembly of Fibronectin into Fibrils. Fibronectin is assembled into fibrils via an integrin-dependent mechanism. Integrins transmit actomyosin forces to soluble FN, which stretches the FN to reveal cryptic FN-FN binding sites. Binding of soluble FN to the stretched FN creates new integrin binding sites that facilitates the assembly of an insoluble FN fibril. Created with Biorender.com.
Active TGF-β ligands signal through a complex of cell surface receptors, comprising type I (TβRI), type II (TβRII), and type III (TβRIII) serine/threonine kinase receptors. During TGF-β signal transduction, a TGF-β ligand first binds to either a TβRII dimer or a pre-formed complex of a TβRI dimer and an RII dimer on the extracellular cell surface [
23,
24,
25]. Ligand-bound TβRII then initiates recruitment of TβRI to form a heterotetramer complex. Constitutively active TβRII then phosphorylates the catalytic domain of TβRI on the cytoplasmic side, initiating downstream signal transduction [
23,
24]. TβRIII, also known as beta-glycan, can enhance the binding of TGF-β ligands to TβRII and is capable of complexing TβRI & II receptors together, serving as a co-receptor to increase TGF-β signaling in certain cell types [
26,
27,
28].
While TGF-β2 has been implicated in fibrotic processes and TGF-β3 has been primarily identified as playing a role in physiological processes such as embryonic development, the TGF-β1 isoform has been characterized in the greatest detail in disease states [
29]. TGF-β1 demonstrates the highest systemic abundance in the human body, with increases in expression associated with poor breast cancer prognosis and metastasis to bone, lymph, and pleural tissues [
30,
31,
32]. It has also been implicated in Parkinson’s disease [
3], fibrotic diseases [
33], and osteogenesis imperfecta [
7]. Each TGF-β isoform exhibits differences in ligand activation and receptor binding. TGF-β2 induces signals with 100–1000-fold less potency than TGF-β1 and TGF-β3 in cells that lack the noncatalytic TGF-βRIII (beta-glycan) co-receptor [
23] and requires a preformed complex of TGF-βRI & TGF-βRII for sufficient ligand-receptor complexing [
25,
27]. TGF-β2 also lacks the typical integrin adhesion motif, RGD, and instead is only able to undergo integrin-mediated activation via an alternative recognition motif that is only recognized by integrin αvβ6 [
34,
35]. TGF-β3 has been shown to have a slightly greater affinity for TGF-βRII [
23] than TGF-β1. TGF-β1 and 2 are known to promote scar formation during wound healing, while TGF-β3 promotes scar-less wound healing, indicating potentially divergent downstream signaling from TGF-β3 [
36,
37].
TGF-β signaling induces multiple downstream signaling events. In canonical TGF-β1 signaling, SMAD proteins are phosphorylated by the cytoplasmic domain of TβRI. SMAD2 and SMAD3 are the primary phosphorylation targets; these complex with SMAD4 and translocate to the nucleus to regulate target gene expression [
24,
25,
26,
27]. Noncanonical signaling also relies on signaling downstream of TβRI activity, but instead activates the mediators of the Rho family, NF-ΚB, Ras, MAPK, and PI3K signaling pathways to direct cytoskeletal rearrangement, protein synthesis, proliferation, and apoptosis [
38,
39].
1.2. Fibronectin Fibrillogenesis and Regulation
A primary component of fibrotic tissue is fibronectin (FN) [
40,
41], which serves both structural and signaling roles in
de novo tissue morphogenesis. FN fibrils serve as scaffolds for the assembly of a variety of other ECM components, including collagens and elastins, facilitating the formation and maturation of new tissue [
42,
43,
44]. This is an important point in fibrosis research: studies have shown that cells lacking FN are unable to assemble Collagen I fibrils in vivo [
45,
46,
47]. This would suggest that inhibition of FN should also inhibit Collagen I assembly and, as such, serve as a novel target for fibrotic treatment. Indeed, treatment with an FN inhibitor in a unilateral ureteral obstruction (UUO) mouse model of kidney fibrosis demonstrated a significant reduction in Collagen I, Collagen III, and fibronectin in UUO kidneys [
48].
In addition to serving roles as a structural element and a scaffold for Collagen I, assembled FN fibrils are also capable of binding upwards of 40 growth factors, including platelet-derived growth factors (PDGFs), fibroblast growth factors (FGFs), and of particular interest, latent TGF-β1 [
10,
15,
41,
49], making it a crucial regulator of cellular signal transduction in fibrotic foci. As such, FN fibrils not only dictate the mechanical structure of the fibrotic ECM, but also play a key role in regulating signaling cues to resident cells.
FN is secreted as a soluble dimer that binds primarily to cell-surface integrin receptors, most notably integrins α5β1 and αvβ3. These integrins transmit actomyosin contractile forces to the FN molecule, which stretches FN into an open conformation that facilitates FN-FN binding [
42,
43]. This cycle of stretching to expose new binding sites and subsequent incorporation of soluble FN leads to the assembly of insoluble fibrils in a process known as fibrillogenesis [
42,
43,
50] ().The polymerization of FN into insoluble fibrillar networks serves as a scaffold for cellular adhesion, migration, and signaling, allowing for the subsequent association of other ECM constituents like collagens and elastins [
43,
49,
51]. Further processing of FN fibrils includes both cleavage by proteolytic enzymes, which results in soluble fragments that exert their own cellular signaling function [
52,
53] and cross-linking by matricellular enzymes to reinforce attachments to both other FN fibrils and other ECM components. Both cleavage and cross-linking of FN fibrils dictate the bulk mechanical properties of the local ECM (reviewed in [
43]).
1.3. Role of Integrins in FN Fibrillogenesis and TGF-β1 Activation
A common thread between TGF-β1 activation and FN fibrillogenesis is that both require binding to transmembrane integrins and subsequent transmission of actomyosin-based contractile forces. For FN fibrillogenesis, both α5β1 and αvβ3 integrins bind to the RGD sequence on the 10th Type III domain of FN [
54]. Transmission of actomyosin forces via these integrins stretches FN from a compact conformation to expose cryptic FN-FN binding sites that facilitate FN fibril formation [
55]. While both α5β1 and αvβ3 integrins bind to FN, fibrillogenesis is primarily driven via α5β1 integrins [
43]. For TGF-β1 activation, αv-containing integrins bind to the RGD sequence on the latency-associated peptide (LAP) of TGF-β1. Transmission of actomyosin forces via these integrins stretches LAP to release the active TGF-β1 ligand, allowing it to bind to TGF-β receptors and facilitate downstream signaling [
56].
1.4. Targeting Integrins and TGF-β1 Therapeutically
The clinical significance of FN fibrillogenesis, TGF-β1 signaling, and integrin signaling in fibrotic diseases and cancer is evident by the vast number of clinical trials that have attempted to target these pathways [
2,
3,
4,
57,
58,
59,
60,
61,
62]. Over 90 integrin-based therapeutics are under investigation, but only seven have been successfully marketed [
58]. Targeting integrins has been proposed as a therapeutic approach to cancer [
59,
63] including glioblastoma [
64], inflammatory bowel disease [
57], cardiovascular disease [
60], dry eye disease [
60], idiopathic pulmonary fibrosis [
62], and scleroderma fibrosis [
65], amongst others. Similarly, targeting TGF-β has been a therapeutic target for a host of diseases, including cancer [
2,
4,
5], Parkinson’s disease [
3], and osteogenesis imperfecta [
7].
1.5. Underexplored Aspects of TGF-β-Integrin-FN Signaling
The substantial number of clinical trials that have targeted either integrins or TGF-β1 indicate the significant role and pervasiveness of these pathways in fibrotic diseases; however, the large number of failures of these clinical trials [
59,
63,
66] suggests an incomplete understanding of their complex signaling and crosstalk. Where are the shortcomings in understanding these signaling pathways? Here, we discuss three areas that may lie at the heart of this issue: (i) divergent regulation of TGF-β isoform expression in response to exogenous TGF-β1 depending on cell type; (ii) divergent FN expression and assembly responses to exogenous TGF-β1 depending on cell type and matrix microenvironment; and (iii) downstream integrin signaling in response to TGFβ/LAP binding to integrins.
1.6. Divergent Regulation of TGF-β Isoforms
Exposure to exogenous TGF-β1 often leads to upregulation of endogenous expression of TGF-β isoforms, which has been hypothesized as an autocrine positive feedback loop that drives sustained TGF-β1 signaling and FN fibrillogenesis [
67,
68,
69]. However, this response appears to be cell-type specific. Several studies have shown an upregulation of autocrine TGF-β1 in MDCK renal epithelial cells, MCF10 human mammary epithelial cells, and HK-2 human renal proximal tubular epithelial cells during TGF-β1-induced EMT [
68,
70,
71], while others have shown no significant increase in autocrine TGF-β1 in MCF10A human mammary epithelial cells in response to pathological levels of exogenous TGF-β1 [
69]. A possible theory to explain this discrepancy may be that TGF-β1 activity may predominantly be upregulated by increased accessibility of previously deposited latent TGF-β1 embedded in the ECM, which becomes increasingly accessible via upregulation of actomyosin contractility and matrix remodeling after initial TGF-β stimulation. Other studies have shown an upregulation of endogenous TGF-β2 in response to TGF-β1 in both MCF10A human mammary epithelial cells [
69] and HCC 1954 human breast cancer cells [
18], while other studies have shown a downregulation of TGF-β2 in response to TGF-β1 in MDA-MB-231 triple-negative breast cancer cells [
69]. Interestingly, Barney et al. demonstrated that upregulation of TGF-β2 and FN correlated with cell survival in an in vitro model of tumor dormancy [
18], while other studies indicated that TGF-β2 is elevated to significant levels in TNBC [
72,
73].
Divergent regulation of TGF-β isoforms may be regulated through several mechanisms. One avenue may be via posttranscriptional regulation of TGF-β translation. Liu et al. have proposed a mechanism of competitive endogenous FN mRNA expression, in which FN is transcribed solely as a mechanism to competitively bind miRNAs that inhibit TGF-β expression, thus upregulating TGF-β expression [
67]. Another avenue for divergent responses to exogenous TGF-β1 is altered regulation of TGF-β receptors. As with most receptor-ligand pairs, TGF-β signaling is regulated by the internalization of receptors on the surface (reviewed in [
74]). Specific regulation of receptors outside of clathrin-mediated mechanisms may play a key role in regulating divergent responses to TGF-β1. For example, β-arrestin2, which typically binds to 7-Transmembrane (7TM) receptors to drive internalization and inactivation of G-Protein Coupled Receptors, also mediates endocytosis of TβRIII through a similar mechanism to downregulate TGF-β1 signaling [
75]. Collagen-α2β1 interactions downregulate TβR expression in a FAK-dependent mechanism in osteoblasts [
76]; this further points to cross-talk between integrins and TGF-β signaling. The microRNA miR-150 drives expression of TGF-βRII in intra-epithelial lymphocytes to increase responsiveness to TGF-β1 [
77].
1.7. Divergent FN Expression and Assembly
Previous studies have shown that mechanical activation of TGF-β1 requires tethering of the large latency complex to FN fibrils [
10]. This is presumably due to the need for a resistive force on the LLC to facilitate mechanically induced opening of the LAP-TGF-β complex. Another avenue for divergent responses to exogenous TGF-β1 is distinct regulation of FN fibrillogenesis; inhibiting FN fibrillogenesis with a small bacterial wall peptide has been shown to inhibit TGF-β1 signaling [
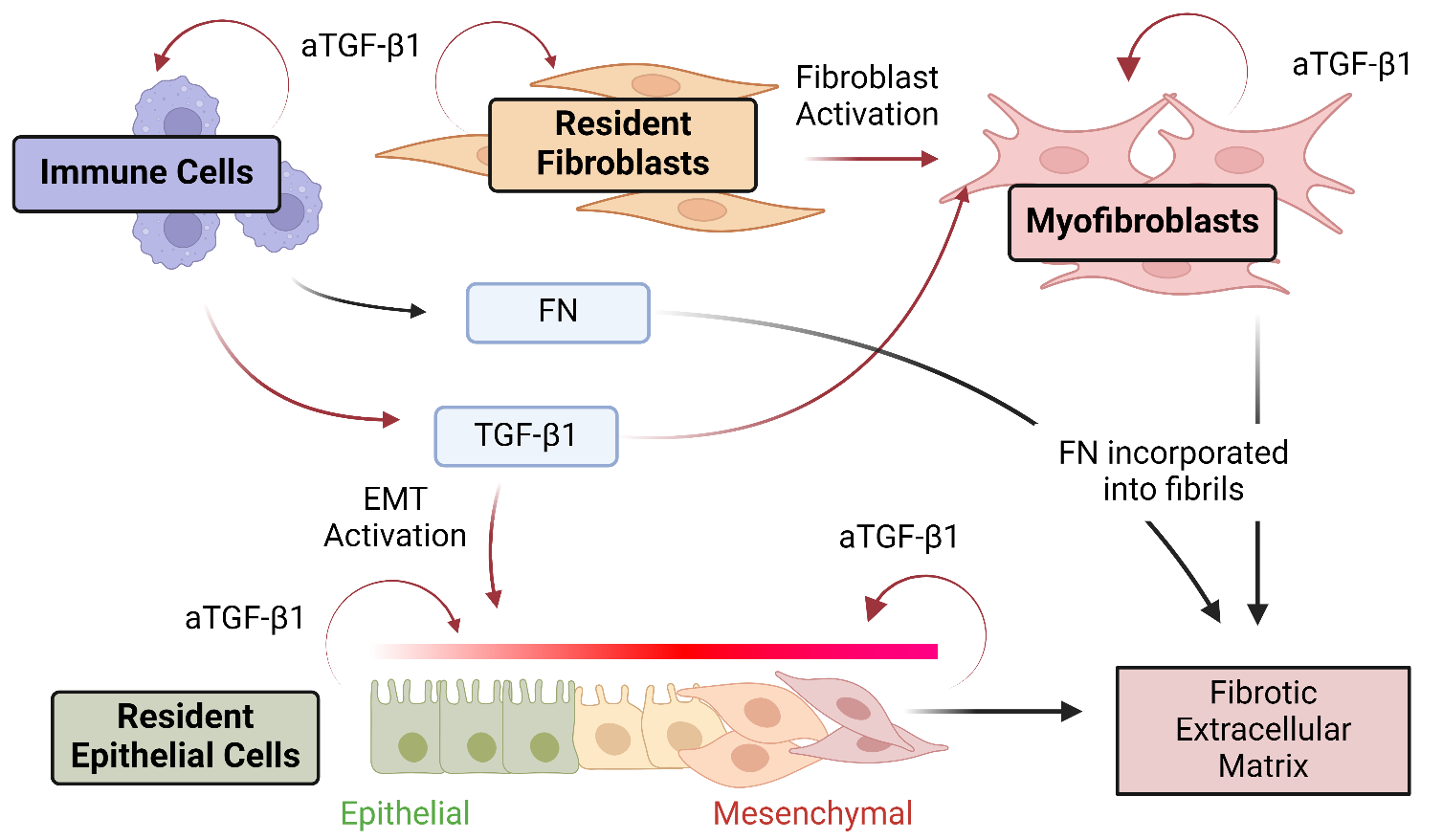
10]. The complexity of this aspect of FN-TGF-β1 crosstalk is amplified by the fact that FN fibrillogenesis requires both the secretion of soluble FN and the assembly of soluble FN into fibrils. This two-step process is not necessarily mediated by the same cell populations: one cell type could upregulate secretion of soluble FN while another could upregulate FN fibril assembly (and a third could upregulate expression of TGF-β1) ().
. Divergent sources of soluble FN, FN fibrillogenesis, and TGF-β1 expression in tissue microenvironments. A critical aspect of the complexity of FN/Integrin/TGF-β1 crosstalk is the potential for multiple cell types/cell phenotypes to contribute to the process. Multiple cell types, including immune cells, resident fibroblasts, and resident epithelial cells, respond to exogenous TGF-β1 by upregulating expression of soluble FN and endogenous, autocrine TGF-β1 (aTGF-β1). Transformed cells, including activated myofibroblasts and epithelial cells that have undergone Epithelial-Mesenchymal Transition (EMT), will assemble FN into fibrils. These fibrils can bind soluble TGF-β1, which can then be activated via integrin-transmitted contractile forces. Figure created with BioRender.com.
Assembly of FN fibrils seems to be more tightly regulated than expression of soluble FN, as there are many cell types and microenvironmental variables that drive increased expression of soluble FN without increased FN fibrillogenesis. Increased expression and secretion of soluble FN has been observed in MDA-MB-231 breast cancer cells [
69], astrocytes exposed to an in vitro stroke model [
78], hepatic stellate cells in a fibrogenesis animal model [
79], HK-2 human renal proximal tubular epithelial cells in response to TGF-β1 [
71], human lung epithelial cells in response to TGF-β1 [
80], and CD14+ monocytes in systemic sclerosis [
81]. These increases in soluble FN expression do not necessarily correlate with increased FN fibrillogenesis: for example, despite significant increases in FN secretion in response to TGF-β1, MDA-MB-231 cells do not readily assemble FN fibrils in 2D cultures [
69], but do assemble fibrils in 3D assays [
82]. Non-malignant MCF10A breast epithelial cells upregulate both FN secretion and assembly [
10] in 2D. Hypoxia upregulates FN secretion in HK2 human kidney cells and A549 human lung adenocarcionma cells, but does not upregulate FN fibrillogenesis [
83]. FN assembly is increased in mouse and human embryonic fibroblasts (3T3 and WI-38) in response to TGF-β1 [
84,
85]. FN assembly is also highly influenced by the abundance of certain ECM proteins in the matrix; culturing cells on laminin-coated surfaces increases FN fibrillogenesis in both MCF10A human epithelial cells [
69] and NRK-49F rat renal fibroblasts (unpublished).
There are several potential mechanisms that could explain how cells facilitate FN fibrillogenesis independent of FN expression and secretion. TGF-β1 induces an increase in surface transglutaminase in mouse and human embryonic fibroblasts (3T3 and WI-38), which co-localizes with α5β1 integrins and may play a role in localizing soluble FN to integrins on the cell surface [
84]. TβRIII promotes epithelial cell attachment to FN by trafficking α5β1 integrins to nascent adhesions [
86]; this effect is independent of exogenous β1. Inhibiting α4 integrin disrupts the binding of the EDA domain of FN to integrins; this results in reduced contractility and reduced FN fibrillogenesis [
87]. Note that the EDA domain is not present in all isoforms of FN; it is an alternatively spliced domain that is not present in systemic FN expressed in hepatocytes (“plasma FN”) but is often present in locally expressed FN (“cellular FN”). As such, divergent expression of transglutaminase, TβRIII, α4 integrin, and/or EDA-containing isoforms of FN could play a role in divergent FN fibrillogenesis responses to exogenous TGF-β1.
1.8. Downstream Integrin-LAP Signaling
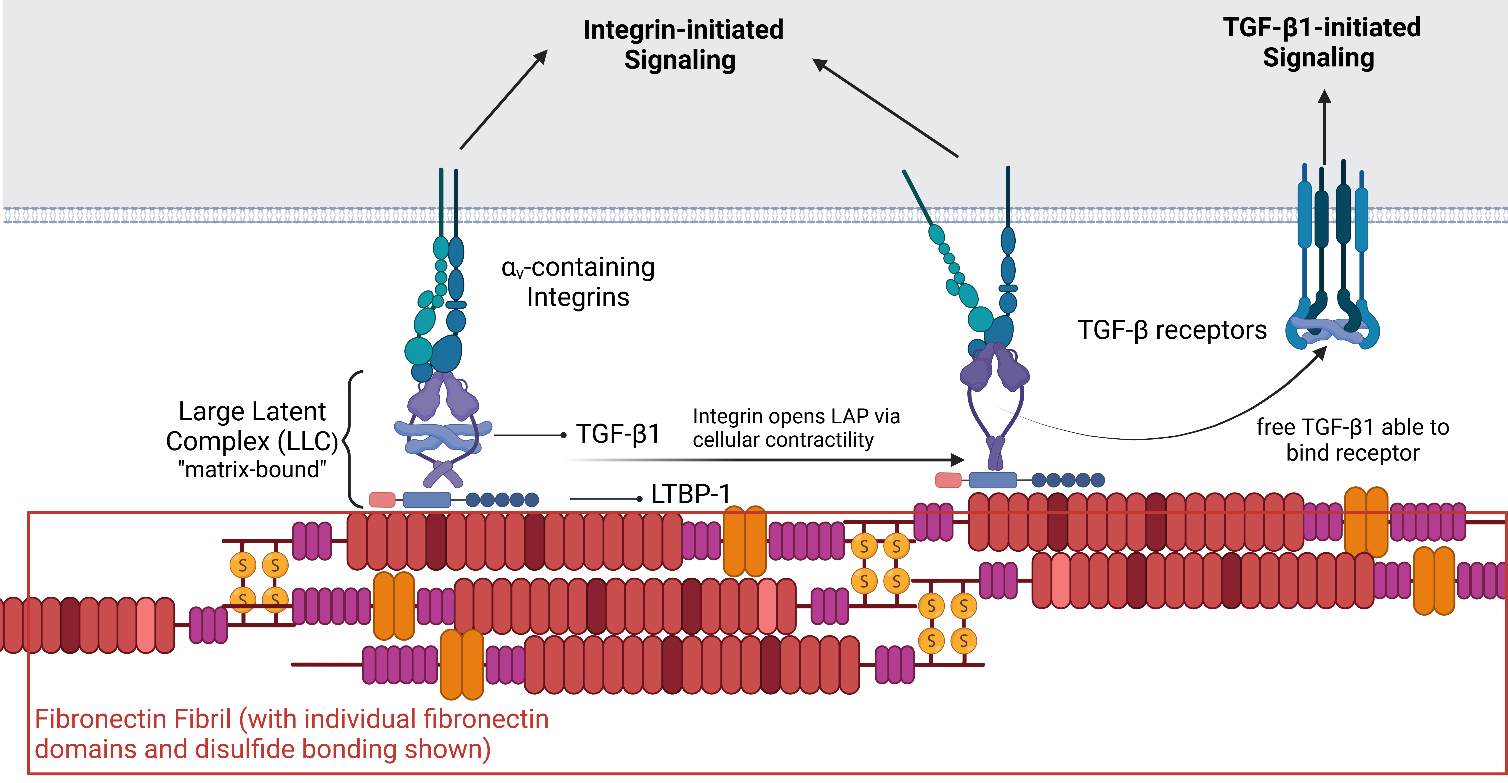
Another potentially important and under-explored avenue of the TGF-β1/FN/Integrin axis is the interaction between the TGF-β1 LAP protein and integrins (). While the importance of this binding is well appreciated and has been explicitly targeted in clinical trials, the focus has been on the role of integrin-LAP binding in facilitating mechanical forces that physically release the active TGF-β ligand. However, LAP-integrin binding may also serve as a traditional receptor-ligand binding event that triggers signaling events downstream of integrin ligation. Results of several studies suggest that this mechanism may play a key role. Studies from our group found that knocking out TGF-β1 resulted in impaired FN fibrillogenesis in MCF10A human mammary epithelial cells, which was not recovered by the addition of soluble TGF-β1. However, addition of the empty LAP protein to TGF-β1 knock out cells did partially recover FN fibrillogenesis [
62]. RGD-binding integrins have the greatest affinity for LAP in its empty conformation (that is, with no TGF-β ligand contained within it), highlighting a potential role in modulating cellular signaling. Prior studies have demonstrated that empty LAP facilitates keratinocyte adhesion [
88] and modulates monocyte chemotaxis and inflammation response [
89].
. Crosstalk between integrin signaling, TGF-β1 signaling, and Fibronectin. Integrins bind to the Large Latent Complex (LLC) that contains the active TGF-β ligand; the LLC can either be soluble or bound to FN fibrils. Bound LLC is stretched via actomyosin forces that open the LLC and release the active TGF-β ligand. This ligand can then bind TGF-β receptors. Integrins bound to soluble LLC, matrix-bound LLC, and/or opened LLC may drive intracellular signaling that augments signaling downstream of the TGF-β1 ligand. Created with BioRender.com.
Writing—Original Draft Preparation, M.M.S., C.A.L.; Writing—Review and Editing: M.M.S., C.A.L.; Project Administration, C.A.L.; Funding Acquisition, C.A.L.
Not applicable.
Not applicable.
Not applicable.
This research was supported by the National Science Foundation via Award CBET-2302580 (M.M.S., C.A.L.).
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.