Idiopathic pulmonary fibrosis (IPF) is a chronic fibrosing interstitial disease of unknown origin, characterized by radiological and histological features consistent with usual interstitial pneumonia (UIP). It is marked by a progressive worsening of dyspnea and a decline in lung function. Both IPF and PPF are comparable because they have poor prognoses with a median survival time from diagnosis of around 2–4 years without antifibrotic therapy. This review shows the main specific characteristics and differences of epidemiology, pathophysiology, clinical and radiological features, treatment, and prognosis of IPF and PPF.
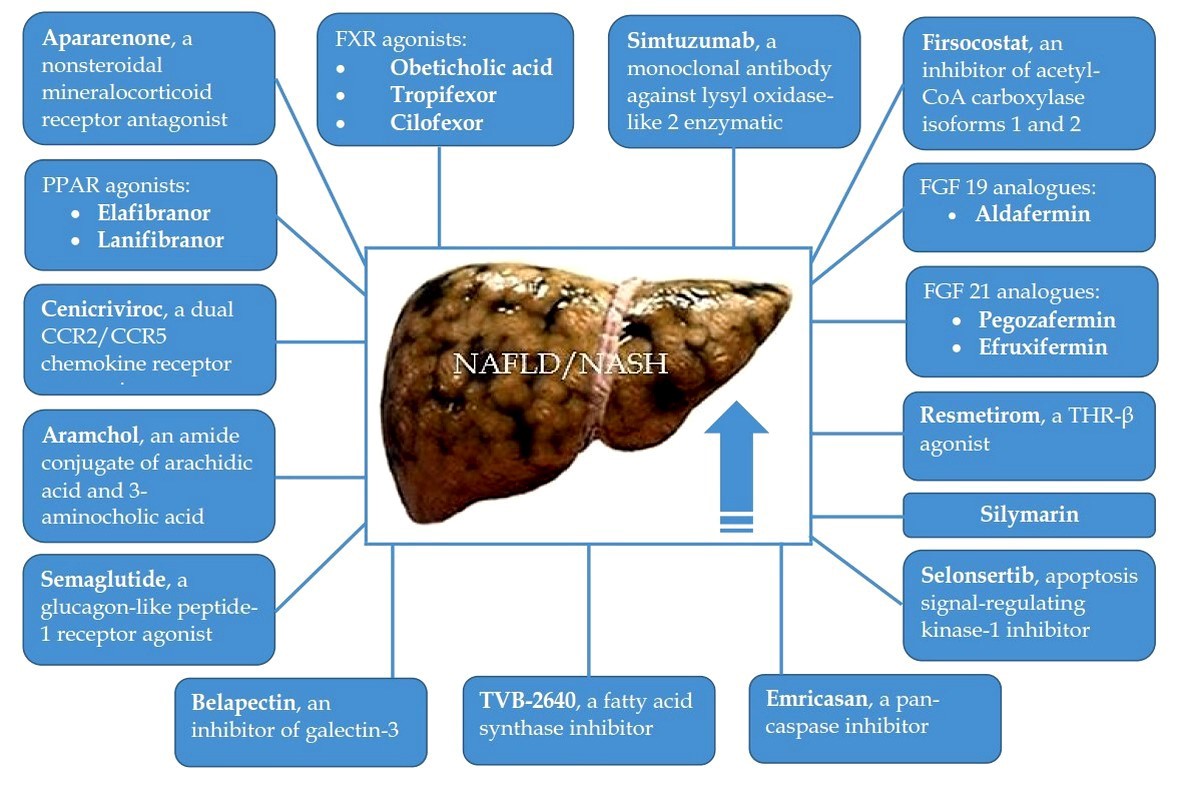
Liver fibrosis (LF) is an adverse event of the natural course of non-alcoholic steatohepatitis (NASH) since its progression leads to the development of liver cirrhosis, which is associated with poor prognosis. In addition, there is evidence that the presence of advanced LF may be a strong independent predictor and risk factor for cardiovascular disease in NASH patients, which is the main cause of their death. Based on the severity of the problem, the study and implementation of drugs for the treatment of NASH-related LF is extremely necessary. The purpose of this review was to describe phase II and III randomized controlled trials (RCTs) evaluating the efficacy and safety of drug therapy for NASH-related LF. To date, the possibilities for pharmacological treatment of NASH-related LF are very limited. However, in recent years, several drugs have been evaluated in NASH patients with LF (F2–3), and in some cases with compensated liver cirrhosis, in large phase II and III RCTs, and they have shown promise. It can be assumed that drugs that have shown efficacy and safety in phase II and III RCTs will be recommended for testing and confirming practical benefits in phase IV RCTs. Besides, an in-depth study of the cellular and molecular mechanisms of NASH-related LF will contribute to the development of new medications, the introduction of which will expand the possibilities of its drug therapy.
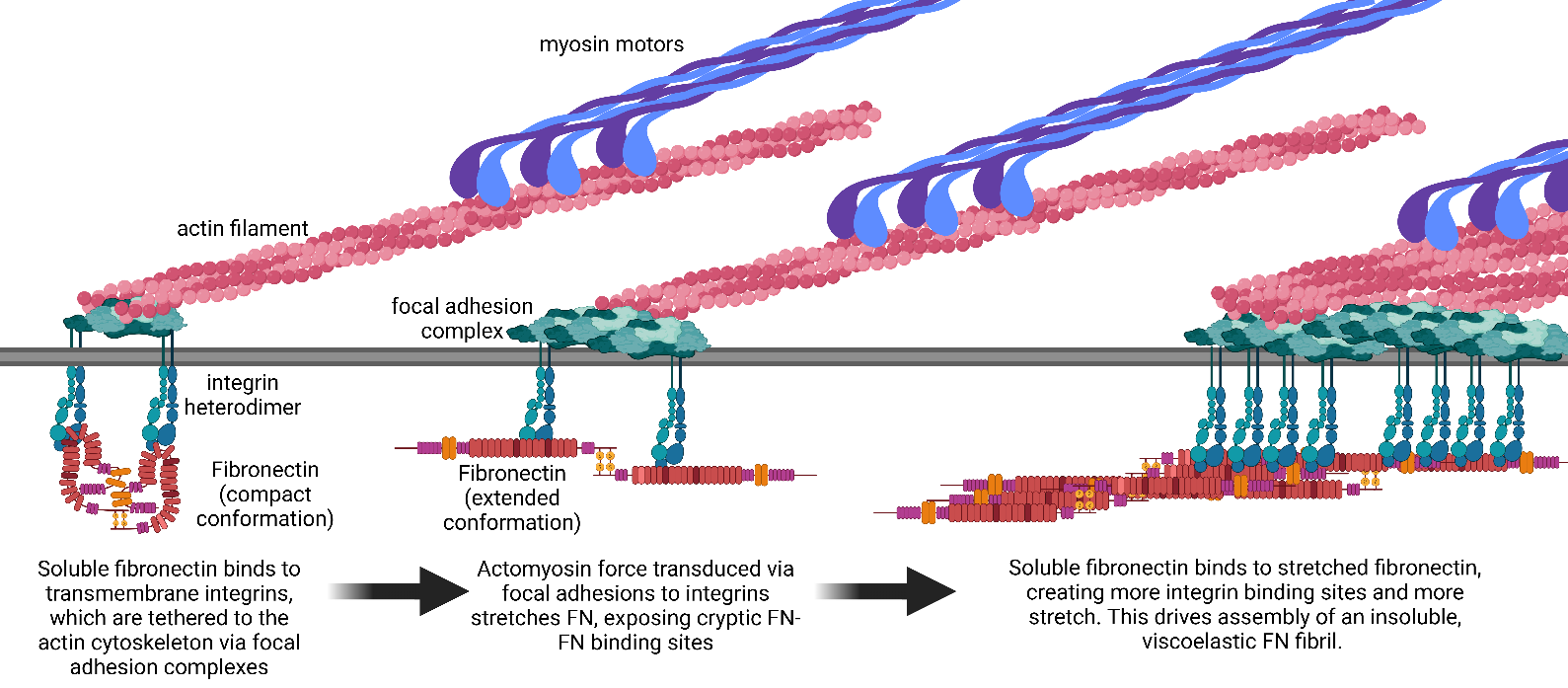
Fibrotic diseases such as pulmonary fibrosis, hepatic fibrosis, chronic kidney disease, and cancer are marked by an excess accumulation of extracellular matrix (ECM). This process involves the assembly of the ECM protein fibronectin (FN) into insoluble fibrils. FN fibril assembly is highly linked with integrin signaling, TGF-β1 signaling, and cellular contractility. This linkage consists of four stages: (i) Integrin binding and contractile forces facilitate the assembly of FN into insoluble fibrils; (ii) assembled FN fibrils bind the large latent complex of TGF-β1; (iii) activation of TGF-β1 from the latent complex requires integrin binding and contractile forces; and (iv) active TGF-β1 increases contractility, integrin expression, and FN assembly. The significance of integrin signaling and TGF-β1 signaling in fibrotic diseases is well-appreciated, as numerous clinical trials targeting integrins or TGF-β1 have been reported. However, despite a clear effort to target integrins and TGF-β1 clinically, the vast majority of these trials have failed or have been terminated. These suggest a potentially incomplete understanding of the synergistic effects of these pathways. Here we present a review of both FN fibrillogenesis and TGF-β1 signaling, as well as current opinions of under-explored areas of crosstalk related to these pathways that may explain why these have not been successfully targeted in many disease states including fibrosis.
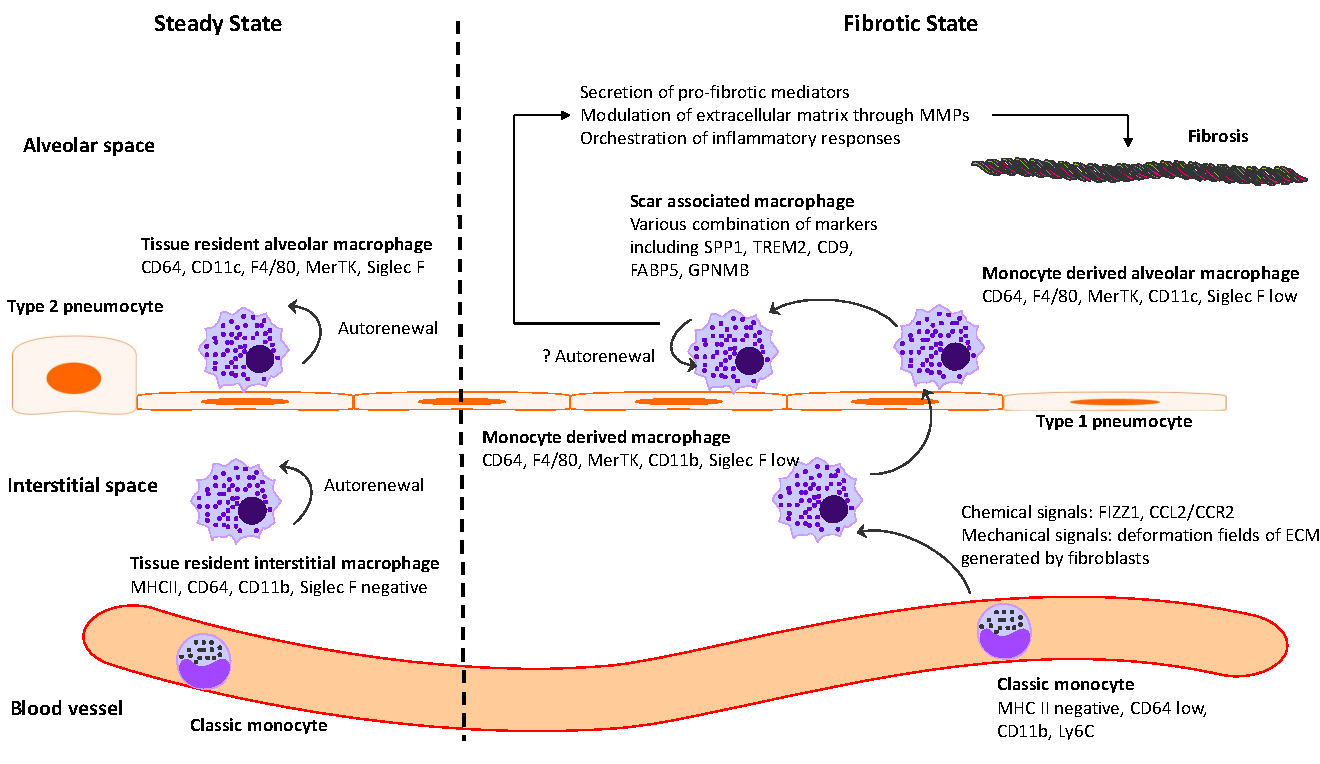
Idiopathic pulmonary fibrosis (IPF) is marked by progressive alveolar destruction, impaired tissue regeneration, and relentless fibrogenesis, culminating in respiratory failure and death. A diverse array of resident and non-resident cells within the lung contribute to disease pathogenesis. Notably, immune cells, both resident and recruited, respond to cues from sites of lung injury by undergoing phenotypic transitions and producing a wide range of mediators that influence, initiate, or dictate the function, or dysfunction, of key effector cells in IPF pathology, such as alveolar epithelial cells, lung fibroblasts, and capillary endothelial cells. The role of the immune system in IPF has undergone an interesting evolution, oscillating from initial enthusiasm to skepticism, and now to a renewed focus. This shift reflects both the past failures of immune-targeting therapies for IPF and the unprecedented insights into immune cell heterogeneity provided by emerging technologies. In this article, we review the historical evolution of perspectives on the immune system’s role in IPF pathogenesis and examine the lessons learned from previous therapeutic failures targeting immune responses. We discuss the major immune cell types implicated in IPF progression, highlighting their phenotypic transitions and mechanisms of action. Finally, we identify key knowledge gaps and propose future directions for research on the immune system in IPF.