Mesophase pitch, a high molecular mass aromatic material produced during the liquid-phase carbonization of thick cyclic aromatic compounds such as petroleum, coal, and naphthalene pitches, is central to the development of advanced carbon materials. This class of material exhibits unique planar, disc-shaped aromatic structures that, under polarized light microscopy, reveal nematic liquid crystals, known as carbonaceous mesophase pitch. With an optically anisotropic texture morphology, mesophase pitch has been pivotal in diverse applications, including the production of carbon fibers, carbon microspheres, needle coke, and carbon foam. Its significance in material science is driven by its abundant source materials, cost-effectiveness, outstanding inherent properties, and high processability [
1,
2,
3,
4].
Despite these advantages, the production of mesophase pitch with optimal characteristics such as high mesophase content and uniform flow domain texture presents significant challenges. One key difficulty is the over-carbonization that occurs as the pitch forms, which leads to an increased softening point and disrupts component homogeneity, complicating further processing [
5]. High-performance mesophase pitch must be engineered to contain a precise molecular structure while maintaining low softening points for practical applications. This complicates further processing and limits the material’s application potential. To mitigate these issues, recent advancements have involved the modification of feedstock and reaction conditions. The inclusion of cycloalkane structures and short-chain alkyl groups has been shown to inhibit over-condensation reactions, reduce the system’s viscosity, and enhance the fluidity of mesophase pitch, thus improving its quality [
6,
7,
8]. Techniques such as co-carbonization processes [
9,
10] and hydrogenation modifications [
11,
12] have been developed to introduce these beneficial features at the feedstock or mesophase pitch formation stage. Furthermore, the employment of supercritical extraction techniques on raw materials to isolate appropriate molecular size components followed by carbonization has proven effective in producing high-quality mesophase pitch with a low softening point [
13,
14,
15].
The process conditions during mesophase pitch formation are also critical in enhancing the pitch content and improving the structure and quality of the final product. Key control factors include reaction temperature, time, and pressure, which must be carefully adjusted based on the raw materials used to promote the formation of mesophase pitch [
16]. Park [
17] demonstrated that a one-stage pressurized heat treatment followed by a two-stage vacuum heat treatment could substantially enhance the properties and yield of mesophase pitch. This process benefits from the retention of aromatic, light components and naphthenic hydrogen, which prevent localized carbonization in the early stages and improve the reaction system’s fluidity for a more homogeneous new component formation [
18,
19,
20]. However, these light components can also hinder the fusion of large polycyclic aromatic in later stages, potentially causing localized over-condensation that adversely affects the quality of the mesophase pitch [
21]. A two-stage vacuum heat treatment process has been designed to effectively remove these isotropic components, thus promoting the fusion of large polycyclic aromatic structures and facilitating the rapid formation of mesophase pitch of the desired size [
22,
23,
24]. Despite these technological advances, the yield of mesophase pitch under optimized conditions remains low, approximately ranging from 20.00 wt.% to 40.00 wt.% [
22,
25], highlighting the need for further research to refine these processes.
The content of quinone insoluble (QI), as the insoluble component in the mesophase pitch, is commonly used to evaluate the properties of the binder bitumen for anodes [
26]. Additionally, the interaction between QI and the fusible component determines the fusibility and optical anisotropy of the entire mesophase pitch [
25,
26,
27,
28]. Therefore, the interaction between the meltable and immiscible components is a key concept for understanding the diverse properties of mesophase pitch. This study aims to address these limitations by focusing on optimizing the two-stage heat treatment processes, which have shown the potential to significantly enhance mesophase pitch yield and improve the quality of the final product. By examining the complex dynamics of phase interactions between QI and soluble components under extended reaction conditions, the research provides a more comprehensive understanding of the role of QI in the mesophase pitch formation process. The findings are expected to offer new insights that could revolutionize the production of mesophase pitch, paving the way for the development of next-generation carbon materials with enhanced industrial applications.
2.1. Analytical Characterization of FCC Slurry
In this study, the basic properties of FCC slurry after catalyst removal were analyzed to assess their suitability for producing high-quality mesophase pitch. The density of the slurry was measured at 20 °C using a precision density meter, adhering to ASTM D4052-16 standards [
29]. Kinematic viscosity was determined at 100 °C using a capillary viscometer, following ISO 3104: 2020 guidelines [
30]. The average molecular weight was assessed through gel permeation chromatography (1260 HT Infinity II, Agilent Technologies, America), and ash content was quantified via gravimetric analysis in compliance with ISO 6245: 2020 [
31]. Elemental composition, including carbon, hydrogen, oxygen, nitrogen, and sulfur, was analyzed using a Varil EL-3 elemental analyzer (Elementar Analysem Systeme Gmbh, Langenselbold, Germany) to ensure accurate profiling. Additionally, the slurry’s hydrocarbon composition was characterized through a SARA (saturates, aromatics, resins, asphaltenes) analysis employing liquid chromatography (UPLC H-Class/xevo G2-S TOF, Waters, Britain) to isolate and quantify each component, highlighting the predominance of aromatic compounds with 3 to 5 rings, which constitute 69.98 wt.% of the raw materials, thereby underscoring its high aromaticity essential for mesophase pitch formation [
32,
33]. This comprehensive analysis is crucial for understanding the physicochemical properties of the FCC slurry, as summarized in , and its potential during liquid-phase carbonization.
.
Physicochemical properties of FCC slurry.
| Properties |
FCC Slurry |
| Density (20 °C) (kg/m3) |
996.60 |
| Kinematic viscosity (100 °C) (mm2/s) |
37.00 |
| Average molecular weight |
389.00 |
| Ash content (wt.%) |
18.00 |
| Carbon residue (wt.%) |
14.39 |
| Elemental analysis |
|
| C |
92.35 |
| H |
6.81 |
| O |
0.01 |
| N |
0.17 |
| S |
0.66 |
| SARA analysis (wt.%) |
|
| Saturates |
12.11 |
| Aromatics |
69.98 |
| Resins |
12.12 |
| Asphaltenes |
5.79 |
2.2. Heat Treatment Protocols for Mesophase Pitch Synthesis
A two-stage heat treatment was carried out in a 500.00 mL high-temperature and high-pressure reactor (Nanjing Bosi Instrument Co., Ltd., Nanjing, China). Initially, 120.00 g of feedstock was placed into the reactor, which was then sealed and rinsed thrice with nitrogen of 99.99% purity. The internal nitrogen pressure was set to 1 MPa. The temperature of the reactor was increased to 425 °C at a heating rate of 5 °C·min
−1 and a stirring rate of 200 revolutions per minute. The start of the reaction was marked from the moment the target temperature was reached (time = 0 h). The first stage of the reaction proceeded under self-generated pressure for durations ranging from 7 to 12 h. Subsequently, the reactor was allowed to cool naturally to 250 °C, and the reaction gases were released.
For the second stage, the product from the first stage was reheated to 425 °C using the same heating and stirring rates. Upon reaching the target temperature, stirring ceased, and the reaction was maintained at this temperature for 1 to 3 h. After this phase, the reactor was again allowed to cool naturally to 250 °C, after which it was opened. The products in the reactor are separated into two phases, upper liquid and underlying solid, and when tested under a polarizing microscope, the liquid phase is completely isotropic (No QI), and the solid phase is mostly anisotropic. The upper liquid and the underlying solid are collected, weighed, and documented separately [
34,
35,
36,
37,
38]. In the reaction, the raw materials are completely converted into gas, liquid, and solid. Therefore, the gas weight can be determined by the mass difference method. The calculation of total yield, solid yield, liquid yield, and gas yield are shown in Equations (1)–(4). The specific reaction times for each stage of the heat treatment, which varied by sample, are detailed in .
Wr: Raw material weight (g)
Ws: Solid weight (g)
Wl: Liquid weight (g)
Wg: gas weight (g)
Yt: Total yield (wt.%)
Ys: Solid yield (wt.%)
Yl: Liquid yield (wt.%).
.
Reaction times for mesophase pitch samples.
| Reaction |
One-Stage Heat Treatment |
Two-Stage Heat Treatment |
| Samples |
S1 |
S2 |
S3 |
S4 |
S5 |
M2.1 |
M2.2 |
M2.3 |
M3.1 |
M3.2 |
M4.1 |
| Stirring (h) |
7 |
9 |
10 |
11 |
12 |
9 |
9 |
9 |
10 |
10 |
11 |
| Non-Stirring (h) |
0 |
0 |
0 |
0 |
0 |
1 |
2 |
3 |
1 |
2 |
1 |
2.3. Physicochemical Characterization of Mesophase Pitch
The physicochemical characterization of mesophase pitch samples began with Soxhlet extraction to separate the fractions based on solubility:
n-heptane soluble fraction (HS), heptane-insoluble/toluene-soluble fraction (HI-TS), toluene-insoluble/quinoline-soluble fraction (TI-QS), and quinoline-insoluble fraction. The anisotropic texture of the mesophase pitch samples was then examined using polarizing optical microscope (Leica DM2700P, Leica Microsystems, Wetzlar, Germany). Thermal decomposition behaviors were analyzed under a nitrogen atmosphere at a 100 mL/min flow rate, with temperatures ranging from 20 to 800 °C at 5 °C/min heating rate, using a Thermogravimetric analyzer (TGA) (Labsys Evo, Setaram Labsys, Caluire, France). For each test, 5 g (±0.1 g) of the sample was weighed and passed through a 200-mesh sieve. The experiment was repeated three times for each sample to minimize experimental variability. Carbon and hydrogen contents were determined using a Varil EL-3 elemental analyzer (Elementar Analysem Systeme Gmbh, Langenselbold, Germany). Further, the functional groups and structural changes within the solid phase products were investigated via Fourier Transform Infrared spectroscopy (FTIR) (Tensor II, Brook, Hamburg, Germany). The crystalline structures were assessed by X-ray diffraction (XRD) (Smartlab-9, Rigaku Corporation, Tokyo, Japan) with a RIGAKU (D/max-RB). The test used Cu source radiation and was taken in the range of 2 to 80 with a step size of 0.2° at a scan rate of 1°/min. Equations (5)–(8) are used to calculate the data, where
θ is the diffraction angle,
λ (0.15418 nm) is the wavelength of the X-rays used,
β is the half-maximum line width in radian [
39].
d002: Molecular interlayer spacing
Lc: Stacking heights
n: Layer numbers
Og: Orientation degrees.
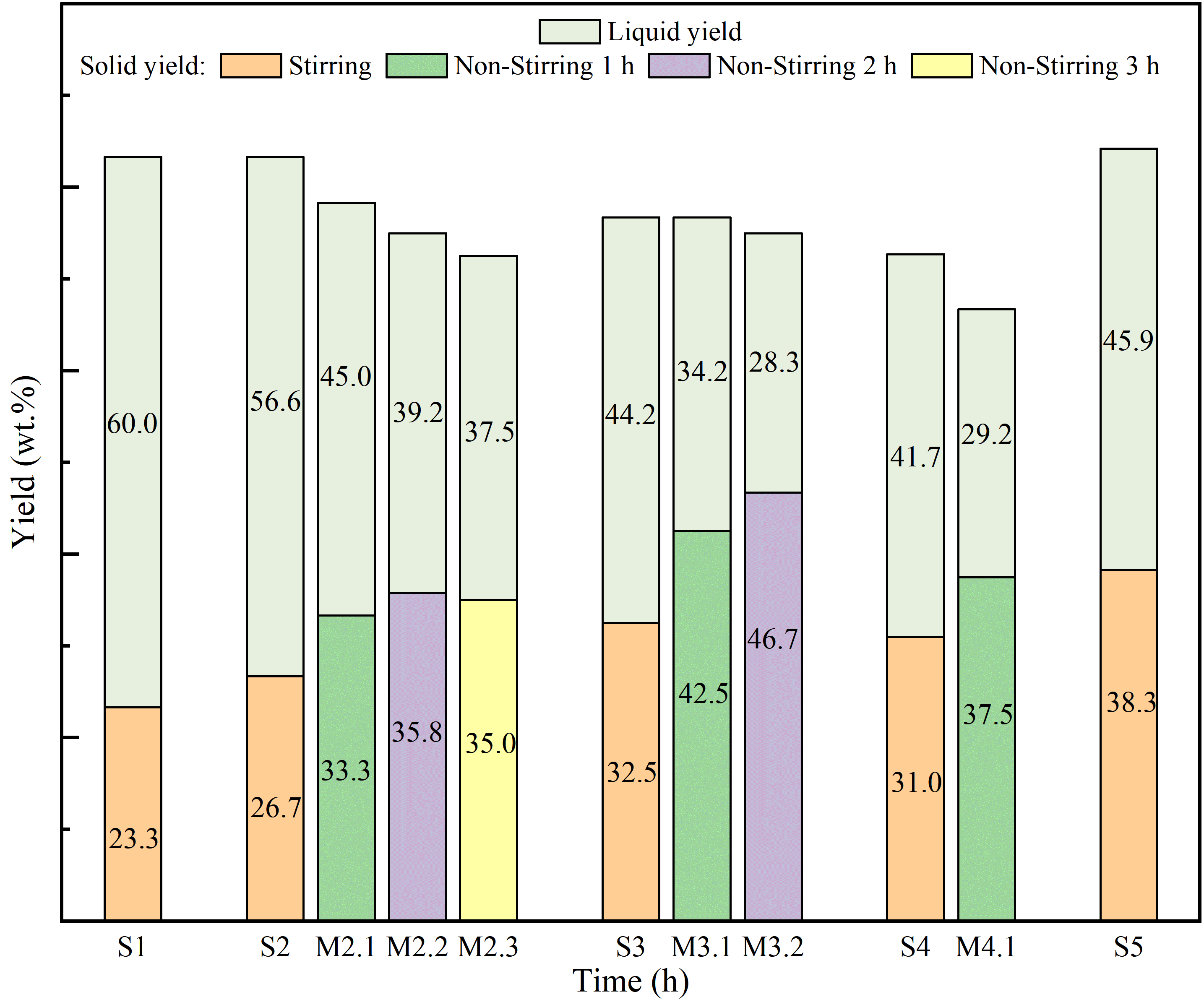
3.1. Impact of Reaction Time on Yield
The variation in yields with reaction time across different heat treatment stages shows distinct trends, as shown in and . Shorter non-stirring holding times at a certain stage (S3–M3.1) notably increase the solid phase yield, in contrast to the yield gains observed with extended reaction durations (S3–S4). This phenomenon can be attributed to the varying concentrations of free radicals at different stages; maintaining reactions at points where free radicals are more concentrated enhances macromolecular radical binding, thus maximizing solid phase production [
40,
41].
.
Solid, liquid and gas product weight under different reaction conditions.
| Product |
S1 |
S2 |
S3 |
S4 |
S5 |
M2.1 |
M2.2 |
M2.3 |
M3.1 |
M3.2 |
M4.1 |
| Solid (g) |
27.96 |
32.04 |
39.00 |
37.20 |
45.96 |
39.96 |
42.96 |
42.00 |
51.00 |
56.04 |
45.00 |
| Liquid (g) |
72.00 |
67.92 |
53.04 |
50.04 |
55.08 |
54.00 |
47.04 |
45.00 |
41.04 |
33.96 |
35.04 |
| Gas (g) |
20.04 |
20.04 |
27.96 |
32.76 |
18.96 |
26.04 |
30.00 |
33.00 |
27.96 |
30.00 |
39.96 |
. Yields under different reaction conditions.
During the initial stage, extending the thermal polycondensation reaction time led to a progressive decrease in liquid phase yield and an increase in solid phase yield, the total yield experienced a gradual reduction. This is because the formation of a new mesophase pitch requires the interplay of pyrolysis reactions, nucleation, and liquid crystallinity coupled with the diffusion of mesogen from the surrounding isotropic phase into the anisotropic phase [
9]. Notably, the rate of increase in the solid phase was not uniform; it increased by 9.20 wt.% from S1 to S3 but showed a marginal decrease between S3 and S4. This irregularity is due to the slower conversion rate of monocyclic and bicyclic aromatic hydrocarbons to polycyclic aromatic hydrocarbons compared to their transformation into anisotropic phases [
42]. As the reaction progresses, extensive consumption of active free radicals prevents the formation of new solid phase components. Instead, when no new components are formed, hydrogen transfer is weakened, and polycyclic aromatic hydrocarbons undergo condensation to form smaller molecules [
43,
44]. This will reduce the weight of the polycyclic aromatic hydrocarbons, resulting in a slight decrease in solid phase yield. Pressure polymerization can effectively incorporate some lighter components in the formation of mesogen [
19,
25], thereby increasing the yield of mesophase pitch. At stage S5, as the pressure increases, the original gas-liquid equilibrium will be disrupted, and the molecules that have turned into the gas phase will return to the liquid phase, thus boosting the total yield.
In the second stage, the most substantial increase in solid phase yield occurred when non-stirring heating was preserved at the stage of S3, where the yield rose by 14.20 wt.%. At this juncture, a significant amount of solid phase product was formed (5.80 wt.%), displaying enhanced condensation activity and a robust capacity for free radical absorption. Under non-stirring conditions, the density gradient promoted increased interactions between free radicals and macromolecules, expediting the formation of anisotropic phases centered around polycyclic aromatic. In contrast, lesser increases were observed at S2, attributed to the lack of generation of large amounts of polycyclic aromatic hydrocarbons and the lower polycondensation activity of the components. The molecules with higher reactivity can break their side chains and release free radicals earlier, while molecules with lower condensation reactivity require a longer time to transform into flat, disc-shaped polycyclic aromatic hydrocarbons [
45]. Therefore, after one hour of holding, yield increases were observed; however, during the second hour, yield gains decelerated markedly due to substantial free radical consumption in the initial hour. Further examination of the reaction processes in M2.1–M2.3 and M3.1–M3.2 illustrates that a similar reaction pattern to the first stage (S2–S4) occurs when free radical content is low, characterized by condensation reactions alongside cleavage to form smaller molecules, thereby slightly increasing or reducing the solid phase yield due to intensified cyclization and deepening carbonation [
46].
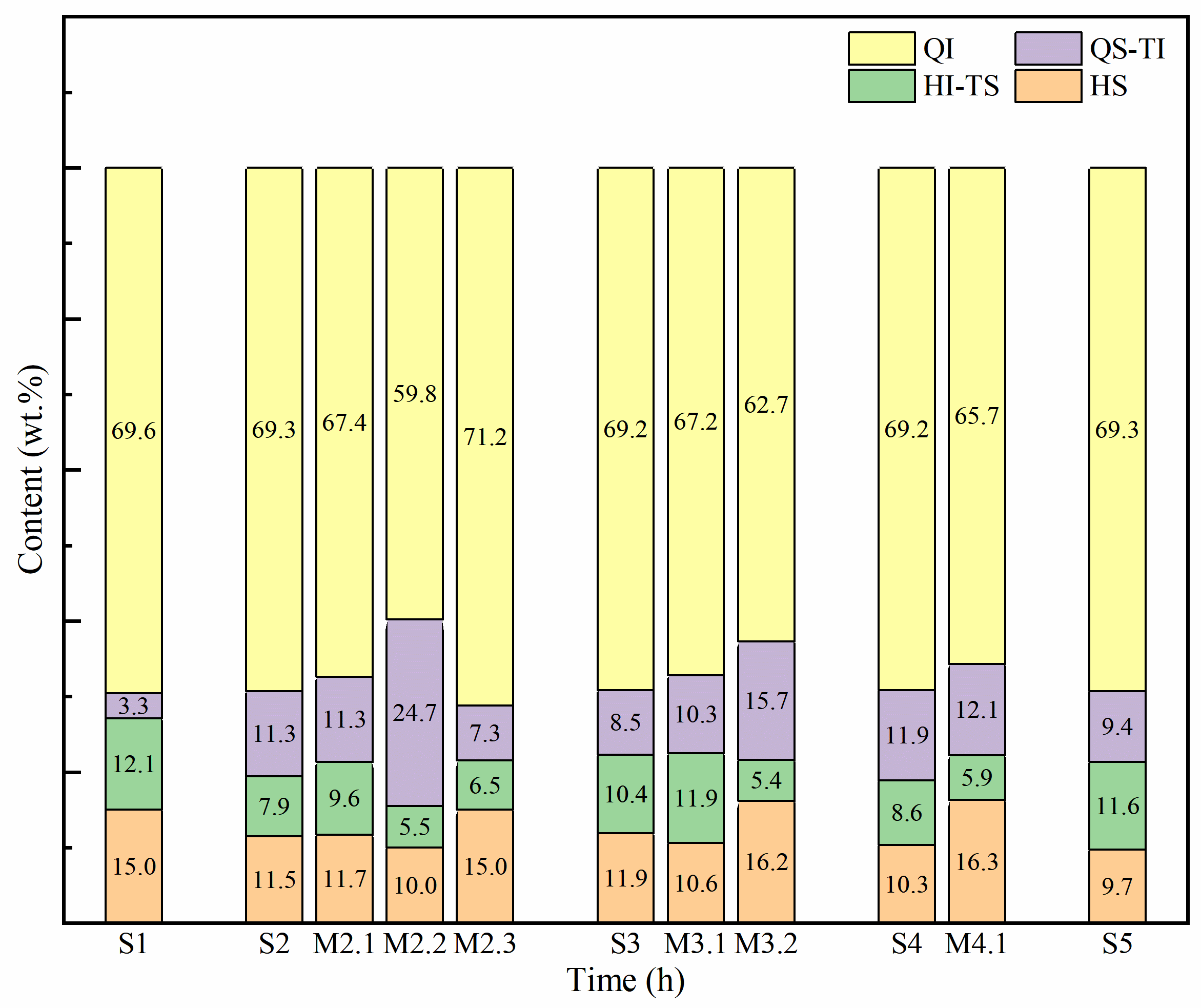
3.2. Impact of Extended Holding on QI Formation
The content of four subfractions in solid phase under different reaction conditions shows distinct trends, as shown in and . In the initial stage of the heat treatment process, aromatic molecules within the reaction system undergo dehydrogenation and condensation, forming polycyclic aromatic. This results in a gradual increase in the QI yield, which stabilizes around 69.00 wt.%. The QI (anisotropy), behaving chemically as a solute and the other three components as solvents, correlates with an increase in solid phase yield. According to reports [
18,
27], the fusible components within mesophase pitch possess a specific quantity of alkyl side chains, and the proportion and properties of these components determine the extent to which the non-fusible components can be fully dissolved. This dynamic equilibrium is crucial for indicating excessive carbonization, as depicted by the trends shown in .
.
The weight of four subfractions in solid phase under different reaction conditions.
| Component |
S1 |
S2 |
S3 |
S4 |
S5 |
M2.1 |
M2.2 |
M2.3 |
M3.1 |
M3.2 |
M4.1 |
| HS |
4.20 |
3.68 |
4.64 |
3.83 |
4.46 |
4.68 |
4.56 |
6.30 |
5.41 |
9.07 |
7.34 |
| HI-TS |
3.39 |
2.53 |
4.06 |
3.20 |
5.34 |
3.84 |
2.37 |
2.27 |
6.07 |
3.02 |
2.66 |
| QS-TI |
0.92 |
3.62 |
3.32 |
4.43 |
4.32 |
4.52 |
10.62 |
3.07 |
5.25 |
8.79 |
5.45 |
| QI |
19.49 |
22.18 |
26.99 |
25.74 |
31.88 |
26.96 |
25.71 |
29.90 |
34.27 |
35.11 |
29.57 |
. The content of four subfractions in solid phase under different reaction conditions.
In the reaction dynamics, HS (isotropic) and HI-TS (isotropic) serve as initial feedstock components. These undergo cleavage, producing small molecule radicals while simultaneously condensing to form TI-QS (partial anisotropic). TI-QS further transforms, breaking down to regenerate lighter components such as HS and HI-TS and condensing into more complex QI [
6]. At a point where no new QI is produced (S4), the existing QI undergoes further dehydrogenation and condensation, leading to a deeper degree of carbonization and a slight decrease in mass.
Extending the holding time beyond optimal parameters can induce structural disparities between newly formed QI and the original components, thereby potentially compromising the quality of the final product [
47]. During the second stage of the heat treatment, non-stirring heat preservation improves the solid phase yield by promoting free radicals to bind more to generate lightweight components, which disrupts the homogeneity of the reaction mixture, leading to fluctuations in the proportion of QI. Although the solid-phase yield exhibits a marginal increase during the second hour of holding, the formation of new QI ceases, reflecting a similar reaction pattern observed between stages S3 and S4. This cessation is attributable to the substantial depletion of macromolecular radicals, resulting from the extensive conversion of TI-QS to QI and the production of lightweight components at the preceding stage, which proves insufficient for new component synthesis [
42,
45].
Prolonged holding times exacerbate these structural differences, further degrading product quality. Conversely, heat preservation at points where the generation of new QI is minimal, such as stage S2, exerts negligible effects on QI yield. This phenomenon is due to the diminished polycondensation activity of the original components, where the promoting effect on the condensation of other components is relatively small [
22,
48].
Notably, the most pronounced augmentation in QI yield is achieved by maintaining heat at a stage characterized by high production of new QI (S3). At this juncture, gravitational forces and enhanced condensation of macromolecular radicals contribute significantly to an increase in molecular weight, thereby increasing the yield of the final product [
49,
50].
3.3. Impact of QI Formation on Anisotropic Phase Textures
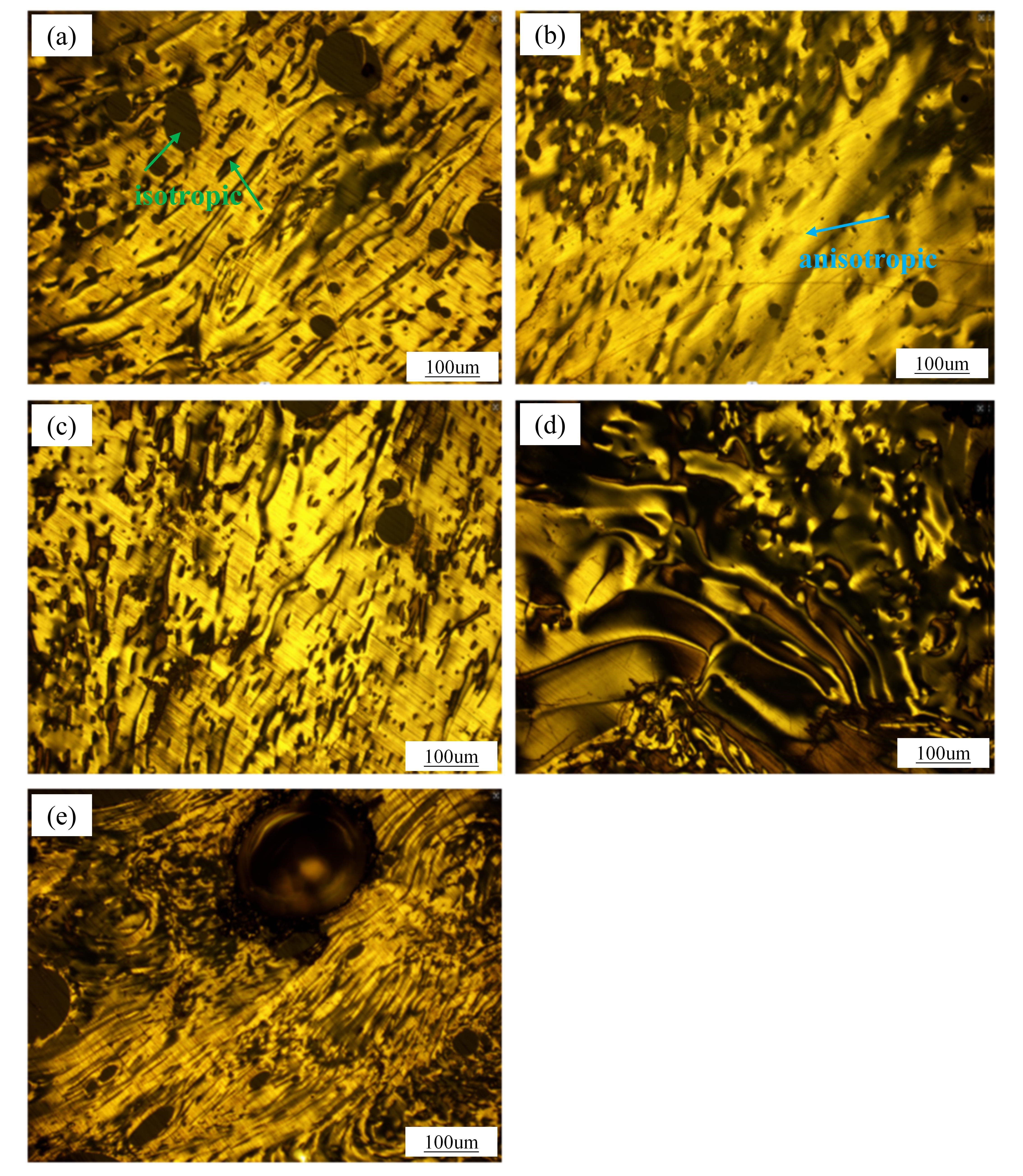
Isotropic absorbs light and shows a completely black color, while anisotropic reflects light and shows a yellow color (as shown in ). During the heat treatment stage, the anisotropic phase texture morphology undergoes significant transformations.
It can be observed in combination with the polarization image in and the ratio of the components of the subfractions in . At the outset (S1), with a minimal amount of newly formed QI, the texture is notably refined characterized by a streamlined optical structure. As the reaction advances to S2, the increase of QI is slow (), and the isotropy is further transformed into anisotropy (, HS + HI-TS → QS-TI + QI), the increase in new components enhancing the texture’s structure (S1 → S2). However, at stage S3, a surge in QI production disrupts this structure due to structural discrepancies between new and original components, reintroducing isotropic characteristics into the solid phase [
51,
52]. Lou [
42] has reported that during the initial stage of carbonization, the formation of TI promotes the aggregation of macromolecules with poor planarity, thereby disrupting the layer orientation and texture of the mesophase pitch. In the absence of new QI formation (S4), the ongoing carbonization process continues to convert isotropic components into the anisotropic phase. Here, the dehydrogenation and condensation processes enhance the structural alignment of the newly formed QI with the original, reducing steric hindrances and leading to the formation of a wide-area streamlined mesophase structure. Nevertheless, by stage S5, the cumulative effects of QI regeneration disrupt the textural balance once more, deteriorating the structure as the reaction cycle progresses.
. Polarized texture of solid phase in the first stage of heat treatment: (<b>a</b>) S1; (<b>b</b>) S2; (<b>c</b>) S3; (<b>d</b>) S4; (<b>e</b>) S5.
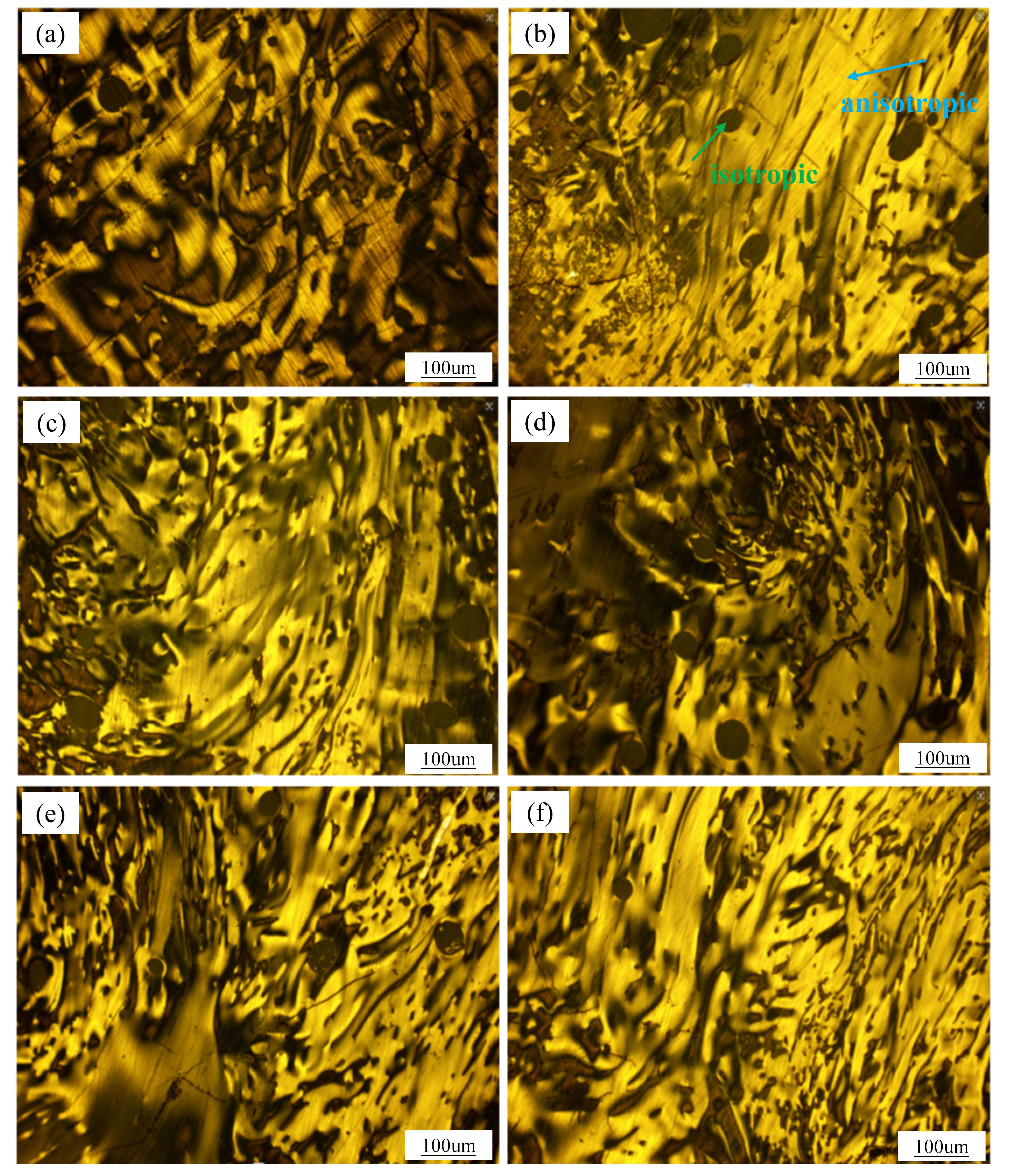
In the second stage of heat treatment, displayed in , the optimal holding time is identified as two hours. Similar to the first stage, structural disruptions occur after one hour due to significant new QI generation. However, the texture morphology improves significantly in the second hour. The substantial consumption of macromolecular radicals during this period halts further QI formation, allowing the polycyclic aromatic more time to align and fuse. This alignment enhances the overall structure of the mesophase [
46]. Nonetheless, due to gravitational forces and stacking effects, newly formed QI tends to accumulate on the surface of the original component. These results in excessive carbonization at the bottom layer, leading to compositional and structural differences across the lengthways of the anisotropic phase [
53].
. Polarized texture of solid phase in the second stage of heat treatment: (<b>a</b>) M2.1; (<b>b</b>) M2.2; (<b>c</b>) M2.3; (<b>d</b>) M3.1; (<b>e</b>) M3.2; (<b>f</b>) M4.1.
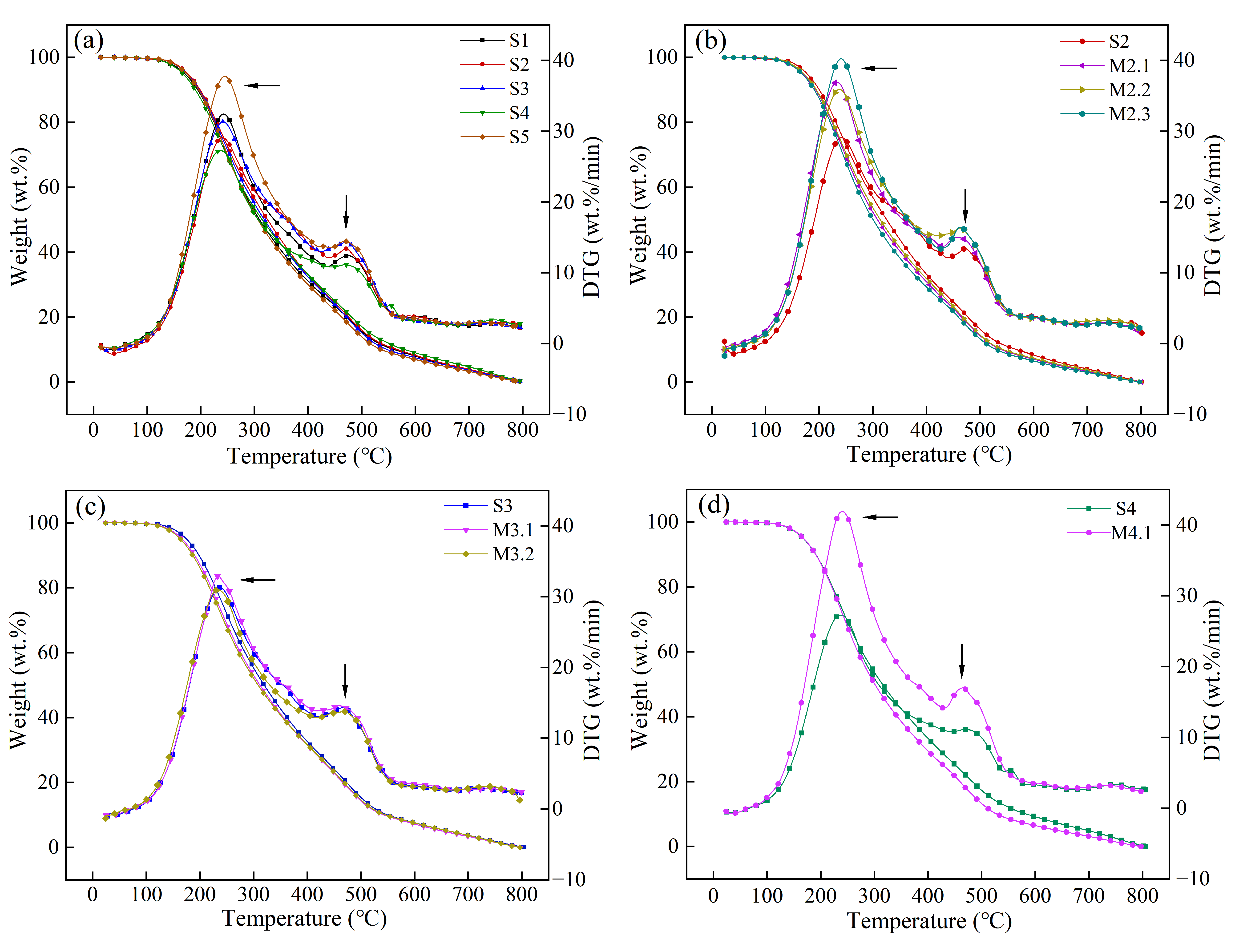
TGA analysis (as shown in ) presents the thermal behavior of solid phase products from various stages of heat treatment. Guo [
54] demonstrated by thermogravimetric analysis that the thermal cracking activity of the four fractions isolated from petroleum residual oils is in the order of saturates > aromatics > resins > asphaltenes. In , the TGA curve features two primary peaks. The first peak, observed around 240 °C, is attributed to the evaporation of light components such as HS, HI-TS, and TI-QS, with the peak intensity reflecting the proportion of light components. The second peak, appearing around 470 °C, relates to the amount of newly formed QI and the extent of carbonization; a higher newly formed QI content enhances this peak due to increased condensability [
53]. Conversely, a lower newly formed QI content and reduced polycondensation activity result in a diminished peak due to decreased reactivity.
Throughout the first heat treatment stage, the first peak exhibits periodic trends with the extension of reaction time, suggesting that the isotropic phase undergoes periodic changes driven by the heat shrink polymerization activities of the molecules. The intensity of this second peak progressively increases with QI accumulation from S1 to S3 but decreases at S4 when the rate of QI formation slows, reflecting reduced reactivity of the newly formed QI. Conversely, as new QI continues to form at S5, the peak ascends once again. During the second stage of heat treatment, after one hour of non-stirring heat preservation, a notable enhancement in both peaks suggests that non-stirring preservation under low pressure effectively promotes solid phase formation. However, the molecular mass remains relatively low with higher molecular activity, resulting in less uniform molecular structures. In the subsequent one hour, the extensive consumption of free radicals limits new component formation, leading to condensation among existing molecules [
18], which increases the overall molecular weight and decreases structural disparities, as reflected by a reduction in both peak intensities. Prolonged holding times, such as seen in M2.3, intensify structural differences due to gravitational effects under non-stirring conditions [
53]. Nonetheless, during the first hour of holding at S3, a significant amount of QI is newly formed, absorbing macromolecular free radicals, which further increases the molecular weight and reduces light component formation. Prolonged holding times lead to a phenomenon similar to other reaction processes, where dehydrogenation and condensation improve the ordering of polycyclic aromatic hydrocarbons, resulting in a more refined texture structure of the mesophase pitch.
. Thermogravimetric analysis of solid phase under different reaction conditions: (<b>a</b>) S1–S5; (<b>b</b>) S2–M2.3; (<b>c</b>) S3–M3.2; (<b>d</b>) S4–M4.1.
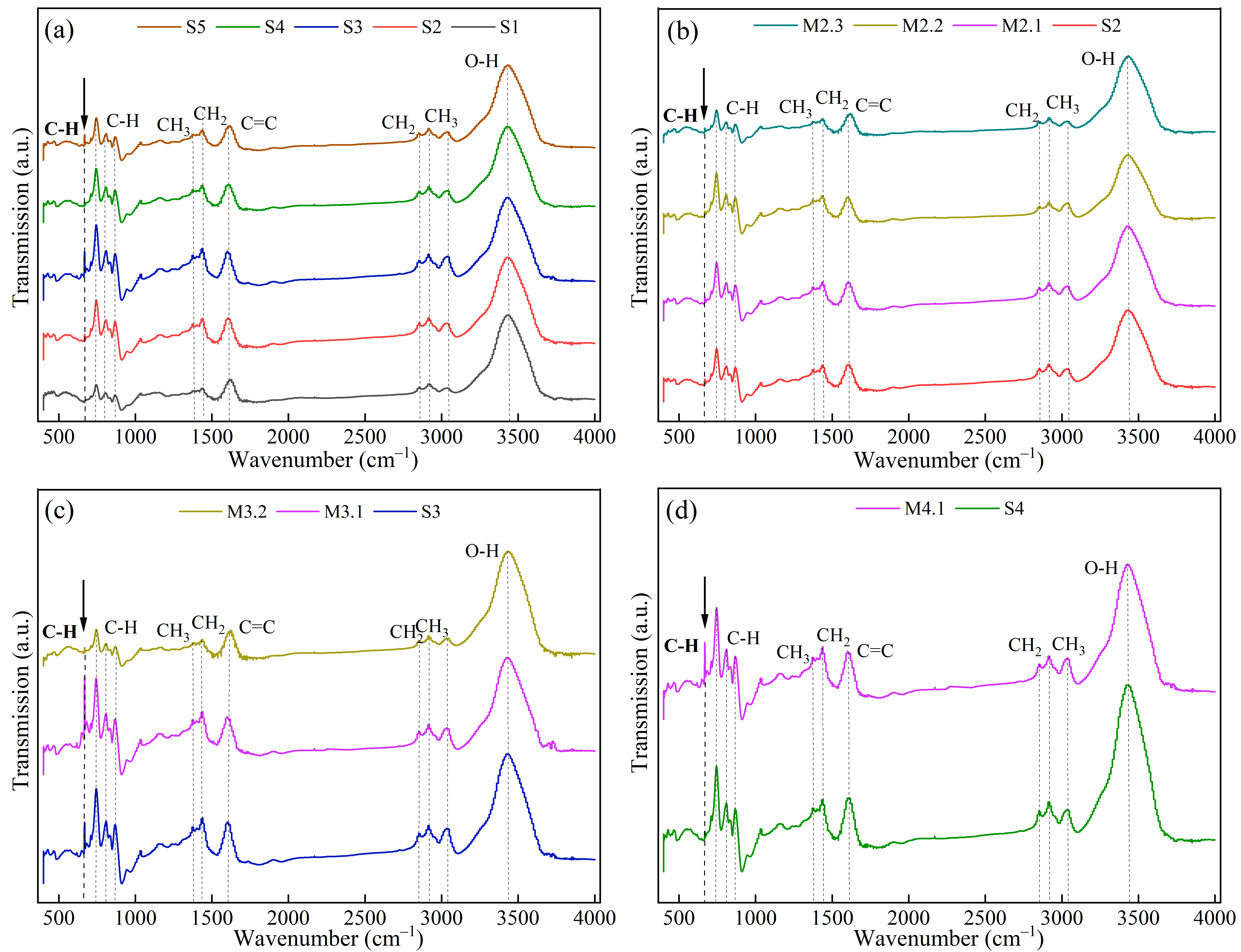
The FTIR spectroscopy analysis reveals detailed vibrational characteristics (As shown in ) indicative of the structural evolution within the solid phase products of mesophase pitch obtained from the first and second stages of heat treatment. Key to the findings, bending vibration peaks of aromatic hydrogen C-H bonds are observed at 750 cm
−1, 810 cm
−1, and 877 cm
−1. The peak at 750 cm
−1, signifying the presence of a kata-condensed aromatic nucleus, suggests a robustly condensed aromatic structure, while the isolated C-H bond peak at 877 cm
−1 indicates a peri-condensed aromatic nucleus. Throughout the reaction stages S1 to S5, the increasing intensity of these peaks highlights a dominant polycondensation reaction, progressively enriching the aromatic content of the product [
55,
56].
Moreover, the FT-IR spectra include skeletal stretching vibrations of aromatic rings at 1610 cm
−1, indicating the increase of aromatic compounds as the reaction progresses. Deformation vibration peaks of CH
3 at 1380 cm
−1 and CH
2 at 1460 cm
−1, along with their respective stretching vibrations at 2850 cm
−1 and 2920 cm
−1, indicating that the solid phase product still contains methyl, methylene, or cycloalkyl-substituted branched chains. Additionally, the appearance of aromatic ring C-H stretching vibration peaks at 3050 cm
−1 (as shown in ) suggests that unsubstituted aromatic hydrogen structures are still present in the solid phase product, indicating a degree of structural integrity in the aromatic framework. However, a significant decrease in the intensity of these peaks suggests thermal cracking and condensation of aromatic side chains, reducing methyl and methylene structures and deepening the condensation process, which ultimately leads to the formation of extensive polycyclic aromatic hydrocarbons [
57].
A critical observation relates to the C-H out-of-plane bending vibration peak at 670 cm
−1, which closely correlates with the quantity of newly formed QI and its polycondensation activity. As new QI is produced, this peak’s intensity becomes pronounced, leading to a deterioration in texture structure. Conversely, when QI undergoes self-polycondensation, the peak intensity diminishes, and the texture structure improves. During the first stage of thermal treatment, the peak intensity increases, reaching a maximum at stage S3, where extensive QI formation occurs. When no new QI forms at stage S4, the original QI undergoes condensation reactions, enhancing the aromaticity and causing the peak to vanish. This disappearance coincides with improvements in the texture structure. However, when a substantial amount of QI forms again at stage S5, the peak reappears, indicating a cyclic pattern of chemical transformations. In the second stage of heat treatment, QI exhibits distinct behavior patterns. After one hour at the S2 holding temperature, the amount of QI generated is comparable to that observed during the first stage, and the peak values are lower, suggesting differences in QI formation under non-stirring conditions. Non-stirring heat preservation enhances the likelihood of condensation between molecules, which modifies the condensation dynamics. The newly formed QI at S2 absorbs less macromolecular free radicals from the lower layers, displaying relatively low polycondensation activity compared to S3. This finding underscores the complex interplay of heat treatment conditions, molecular interactions, and chemical reactions that influence the structural and textural properties of the mesophase pitch.
. FT-IR spectra of solid phase products under different reaction conditions: (<b>a</b>) S1–S5; (<b>b</b>) S2–M2.3; (<b>c</b>) S3–M3.2; (<b>d</b>) S4–M4.1.
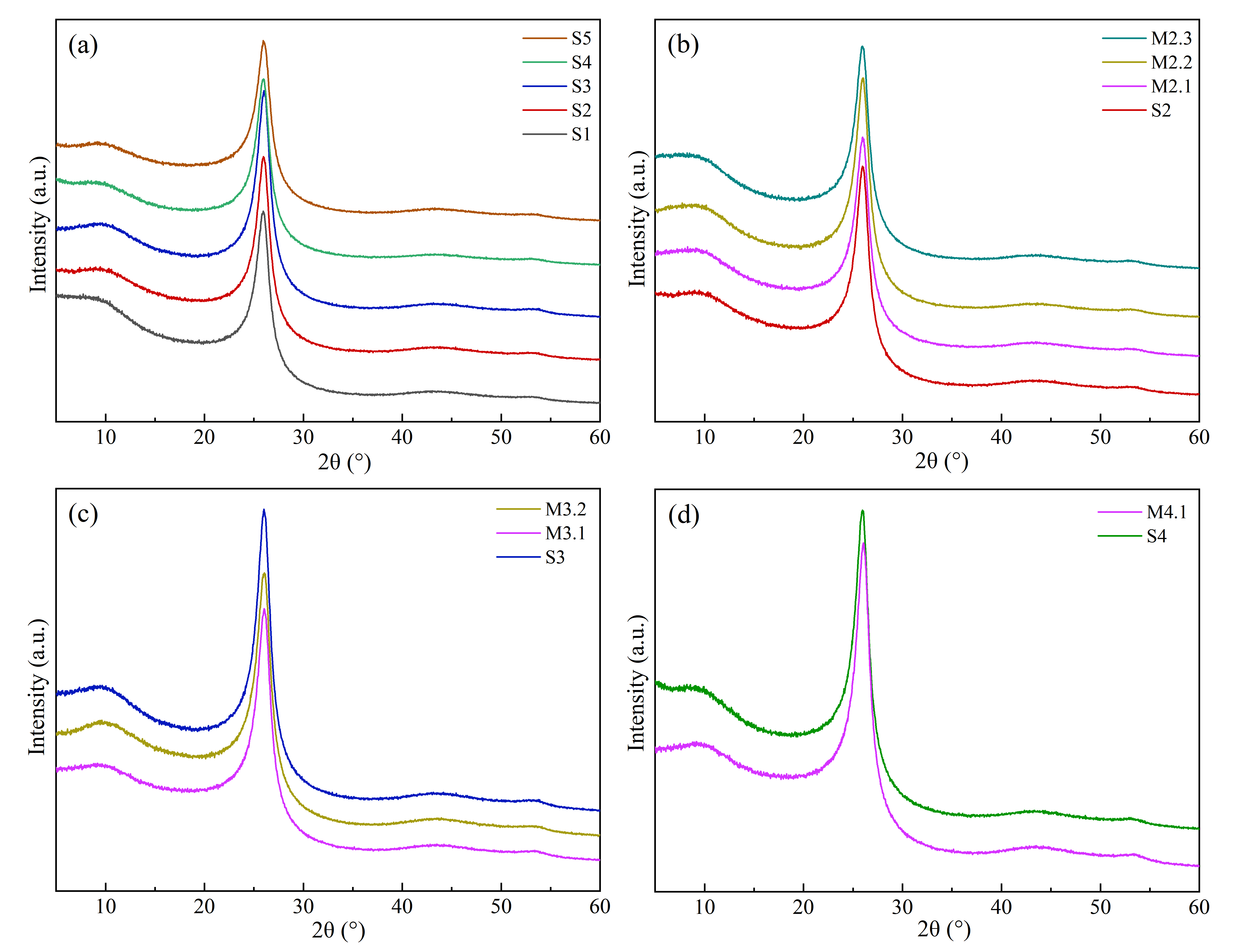
The XRD analysis delineates the evolution of crystal structures within solid phase products throughout various heat treatment stages. The XRD patterns (as illustrated in ) show a pronounced peak at 2θ = 25° and the calculation results are listed in . It confirms a high degree of ordering among polycyclic aromatic, which correlates with the spectral data of molecular interlayer spacing (
d002), stacking heights (
Lc), layer numbers (
n) and orientation degrees (
Og). Lee [
58] points out that the smaller the
d002 and the greater the
Lc, the more ordered the mesophase pitch.
Analysis reveals that the molecular layer spacing and the number of carbon layer stacks vary as the polycondensation reactions intensify. This trend is mirrored by the intensity changes in the C-H out-of-plane bending vibration at 670 cm
−1, providing a marker for QI activity. When QI formation is extensive, the structural disparity disrupts the orderly arrangement of aromatic, reducing the
Lc and
n. Conversely, no new QI formation facilitates closer molecular stacking and improved texture by enhancing the planarity [
25].
. XRD spectra of solid phase under different reaction conditions: (<b>a</b>) S1–S5 h; (<b>b</b>) S2–M2.3; (<b>c</b>) S3–M3.2; (<b>d</b>) S4–M4.1.
.
XRD analysis data of solid phase under different reaction conditions.
| Time (h) |
d002/Å |
Lc/nm |
n |
Og |
H/C |
| S1 |
3.433 |
3.608 |
8.954 |
0.982 |
0.446 |
| S2 |
3.430 |
3.594 |
8.898 |
0.982 |
0.405 |
| S3 |
3.423 |
3.533 |
8.672 |
0.982 |
0.402 |
| S4 |
3.430 |
3.651 |
9.092 |
0.983 |
0.395 |
| S5 |
3.433 |
3.648 |
9.091 |
0.981 |
0.454 |
| M2.1 |
3.426 |
3.578 |
8.833 |
0.982 |
0.407 |
| M2.2 |
3.426 |
3.505 |
8.582 |
0.982 |
0.385 |
| M2.3 |
3.429 |
3.527 |
8.665 |
0.982 |
0.361 |
| M3.1 |
3.420 |
3.534 |
8.667 |
0.982 |
0.405 |
| M3.2 |
3.416 |
3.448 |
8.362 |
0.981 |
0.393 |
| M4.1 |
3.423 |
3.533 |
8.672 |
0.982 |
0.432 |
Throughout the heat treatment phase, the mesophase structure develops rapidly, with polycyclic aromatic initially hindered by long side chains from asphaltenes and heavy gums. As these side chains cleave from S1 to S2, molecular orientation and stacking improve significantly. However, the newly formed QI at stages of S3 obstructs complete aromatic interpenetration, affecting molecular stacking and orientation negatively [
59]. From S3 to S4, the absence of new QI formation allows other molecular aromatization and cyclization, which further consolidates the molecular structure, enhancing stacking. A resurgence in new QI formation at S5 disrupts this order, highlighting the cyclic nature of structural transformations. In the second stage, optimal structural properties are achieved with a two-hour holding period. Initially, increased QI content enhances structural difference, but without stirring, the uniformity of the component distribution suffers, reducing the
Lc and
n. As reaction dynamics slow down, the molecular structure stabilizes, which improves texture and molecular orientation. The reason is that polycyclic aromatic gain sufficient energy to align and interpenetrate more effectively under no-stirring conditions [
22].
The H/C ratio analysis across these stages illustrates a decrease with prolonged reaction time, reflecting increased carbonization and condensation. The first stage shows superior homogeneity and molecular structure, lessening the risk of over-carbonization [
53]. In contrast, the second stage demonstrates that prolonged holding intensifies condensation, intensifying the vertical differences in molecular structure and complicating control in industrial settings.
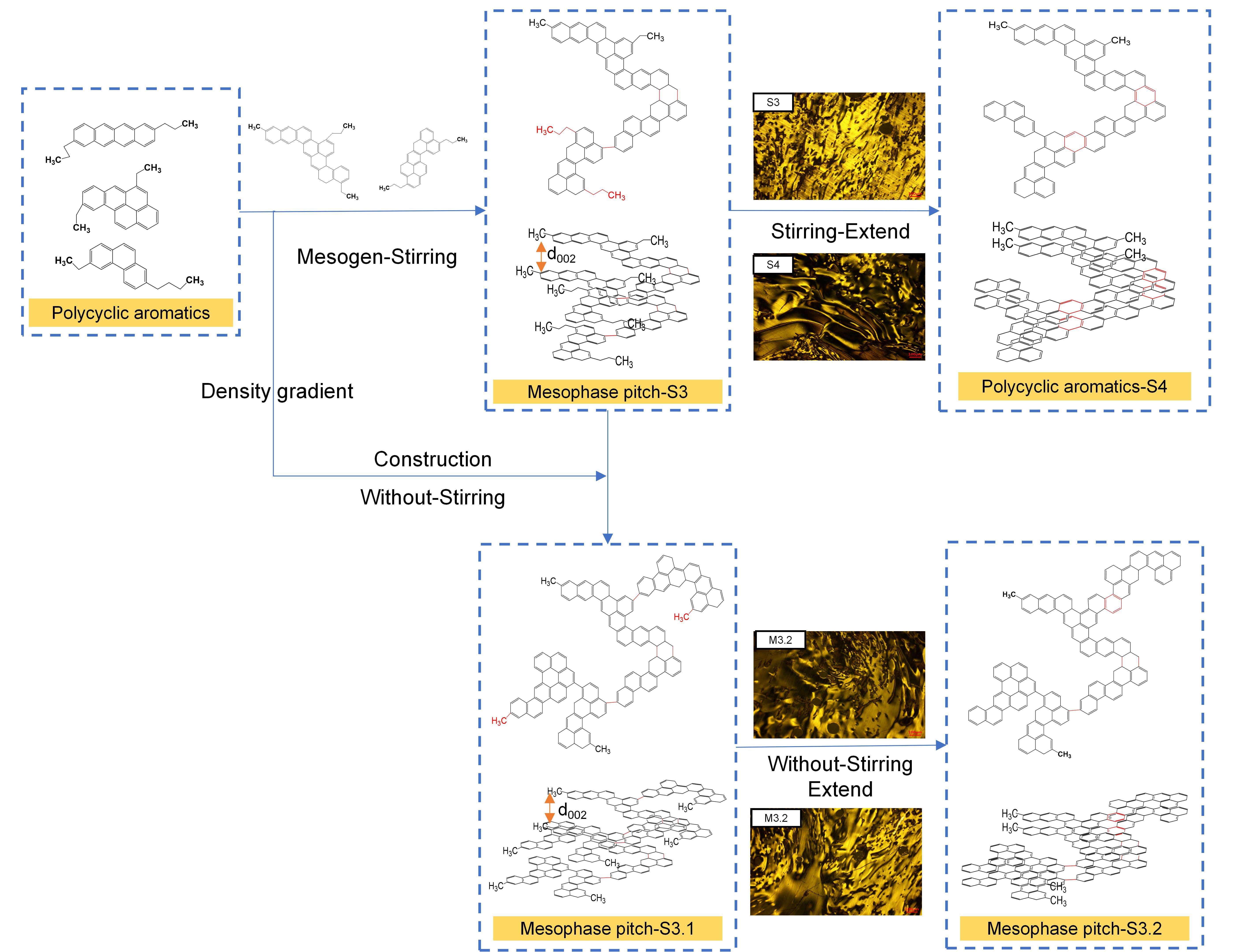
3.7. Formation Mechanism and Structural Evolution of Mesophase Pitch
As shown in , it can be seen that mesophase pitch forms through a two-stage heat treatment of catalytic cracking oil slurry, wherein controlled polycondensation and condensation reactions of aromatic hydrocarbons create an organized structure with anisotropic properties essential for advanced carbon materials. In the first stage, polycyclic aromatics self-assemble into liquid crystal domains. The aromatic hydrocarbons are mostly semi-rigid, have a single alkyl side chain, and the molecular spacing is large, which contains mesogens that are not transformed into anisotropy [
60]. Reaction time is pivotal: extended durations promote the transformation of semi-rigid molecules into rigid ones by enhancing radical interactions and dehyroaromatization. According to Lee [
57], polycyclic aromatic hydrocarbons with semi-rigid structures do not have the same degree of molecular stacking and ordering in the liquid crystal phase as compared to rigid structures. When the increase in QI stopped, the transition from semi-rigid to rigid structures in polycyclic aromatic hydrocarbons led to an enhancement in molecular order. In the second stage, non-stirring heat preservation enables macromolecular condensation to generate larger aromatic structures and promote mesophase molecule integration [
18], refining the anisotropic texture and yield essential for pitch quality.
. Formation mechanism and structural evolution of mesophase pitch.
Thermal and structural analyses reveal that heat treatment lowers the hydrogen-carbon (H/C) ratio. FTIR spectra show increasing aromatic bonds and fewer aliphatic side chains, indicating deep carbonization. XRD analysis further confirms reduced interlayer spacing and increased molecular stacking. Overall, optimized heat treatment yields mesophase pitch with high structural integrity and anisotropy, which is ideal for carbon-based applications.
In this study, mesophase pitch was synthesized using catalytic cracking slurry as the raw material through two-stage thermal treatment methods with and without stirring. The results showed that short-term heat treatment without stirring could effectively increase the solid phase yield from 23.30 wt.% to 46.70 wt.%. The solid phase yield is related to the increase in QI, and the more QI increases, the greater the yield, as can be seen from the stronger peak C-H at 670 cm-1 in FTIR. However, the newly increased QI component disrupted the stability of the solvent system, leading to a decrease in Lc and n, and affecting the quality of the mesophase. When the increase in QI stopped, the C-H peak disappeared. At this time, the QI underwent dealkylation and dehydrogenation condensation reactions, leading to a decrease in intermolecular forces and an increase in Lc and n. As a result, the quality of the intermediate phase was relatively good. Further investigation is necessary to clarify the details of the mechanism of thermal-induced transformation of mesophase pitch. A comprehensive understanding of the dynamics and thermodynamics involved will help develop advanced control strategies that are applicable to different production scales.
H.G.: Investigation, Data curation, Writing—review & editing. Y.L.: Methodology. L.C.: Methodology. P.D.: Methodology. Y.Z.: Writing—editing, Resources. Y.F.: Writing—editing. W.L.: Writing—review & editing, Supervision, Resources, Funding acquisition.
Not applicable.
Not applicable.
The data that support the findings of this study are available from the corresponding author upon reasonable request.
This work was financially supported by the National Natural Science Foundation of China -Petrochemical Joint Fund Key Project (U22B20142), National Natural Science Foundation of China (22078347), Henan Provincial Joint Fund for Science and Technology Research and Development Program(No. 235200810021), Zhongke Technology Achievement Transfer and Transformation Center of Henan Province (2024101), Distinguished Professor of Henan Province (220508001), Foreign Talent Attraction Program of Henan Province (GZS2024023).
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.