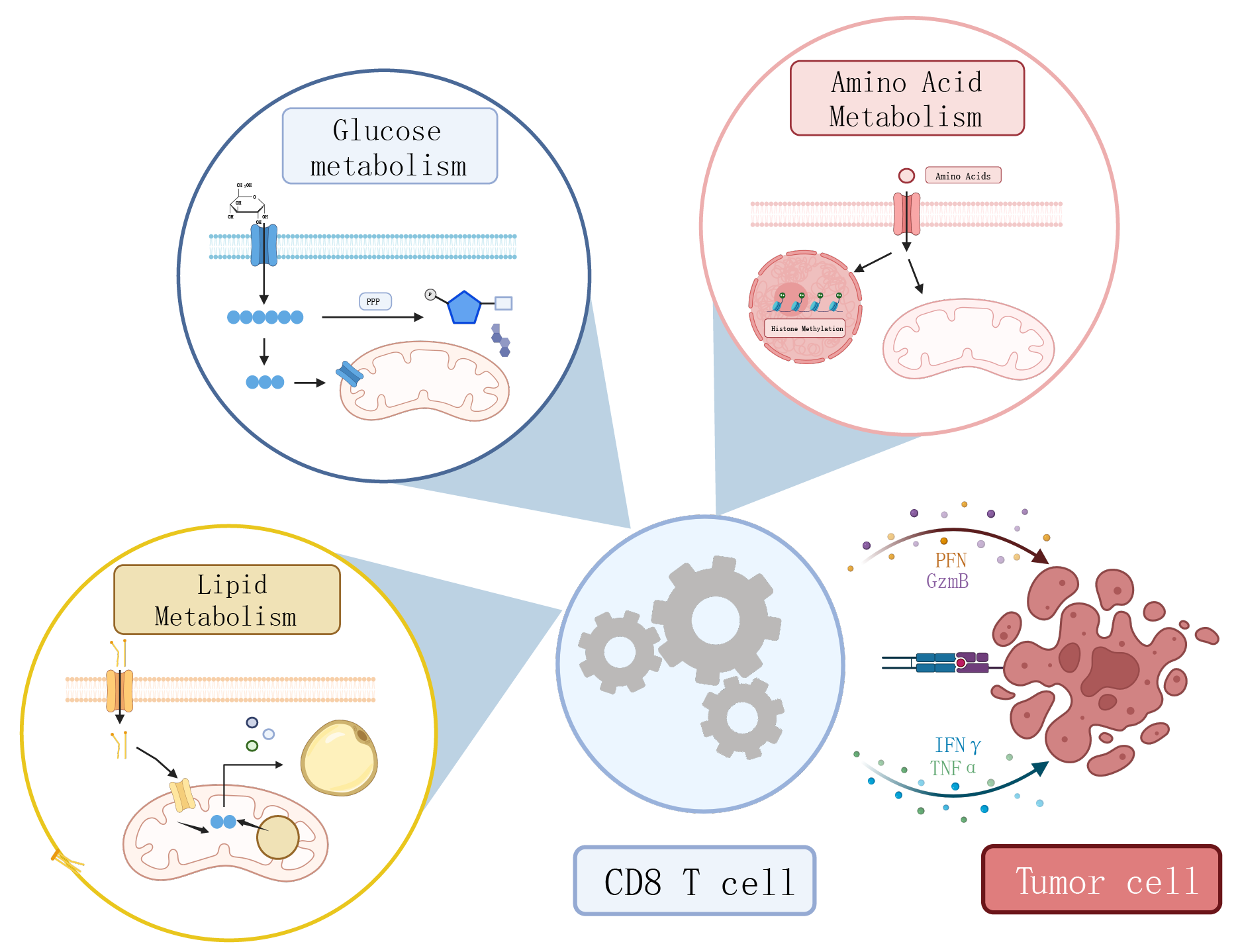
T cell exhaustion is a distinct metabolic and epigenetic state that arises following prolonged antigenic stimulation [
19,
20]. It was first identified in 1993 in lymphocytic choriomeningitis virus (LCMV)-Docile infected mice, where high-dose administration of the virus resulted in dysfunction and, ultimately, clonal deletion of virus-specific cytotoxic T cells without achieving viral clearance [
21]. Subsequent studies confirmed this phenomenon in various human infections, including those caused by human immunodeficiency virus (HIV), hepatitis B virus (HBV), hepatitis C virus (HCV), and in cancer patients [
22,
23,
24,
25,
26]. A pivotal study by E. John Wherry et al. in 2007 provided a comprehensive characterization of exhausted CD8 T cells, defining a common signature that includes upregulation of inhibitory receptors (PD-1, CTLA4, CD244a, and LAG-3), downregulation of naïve/memory-related molecules (CD62L, IL-7R, TCF1, and LEF-1), impaired cell cycling, and a substantial reduction in cytokine production. Metabolically, exhausted CD8 T cells exhibit a marked decrease in the expression of genes associated with different ribosomal subunits and the TCA cycle, underscoring the critical link between metabolic regulation and T cell (dys-) function [
27].
In chronic viral infections and tumors, exhausted CD8 T cells are not a homogenous population but comprise distinct cellular subsets, including precursor-exhausted T cells (T
pex), functional cytotoxic CD8 T cells (T
exint or CTL-like cells), and terminally exhausted T cells (T
ex), as previously described [
28,
29,
30]. Two primary strategies have been identified for enhancing antitumor immunity by counteracting T cell exhaustion: (i) reinvigorating the T
pex population and (ii) supporting the differentiation of CTL-like exhausted T cells while avoiding the accumulation of terminal T
ex subsets. The T
pex subset is the principal population responding to immune checkpoint inhibition (e.g., anti-PD-1 immunotherapy), making its maintenance crucial for sustaining cytotoxic T cell responses in cancer [
31]. Meanwhile, CTL-like cells exhibit potent cytolytic activity and play a central role in antitumor immunity.
Consistent with the observed downregulation of TCA cycle-related genes, multiple studies have demonstrated that suppressed mitochondrial function is linked to an (terminal) exhaustion phenotype of tumor-infiltrating CD8 T cells [
15,
16,
32,
33]. In chronic infection, we and others found that T
ex cells exhibit decreased mitochondrial respiration and enhanced glycolysis compared to T
pex cells. This metabolic shift is tightly correlated with the functional decline of virus-specific T cells [
17]. While T
pex cells maintain robust mitochondrial respiration, T
exint or CTL-like cells rely progressively more on mTOR signaling-dependent glycolysis [
16,
18,
34]. Since T
exint/CTL-like cells persist and dominate during the middle stages of chronic viral infection (e.g., days 14–30 post infection), an overall increase in glycolytic metabolism is observed compared to acute infection, where respiring memory T cells are primarily present at these time points. However, terminally exhausted CD8 T cells exhibit a sharp reduction in both mitochondrial and glycolytic activity, likely due to the cellular degradation of this ‘final-stage’ population.
Various targets have been explored to prevent functional T cell exhaustion: inhibitory receptors, transcription factors, and cellular metabolism. PD-1 and Tim-3 are two examples of inhibitory receptors marking exhausted T cells in mice and tumor patients. Although anti-PD-1 checkpoint inhibition effectively reinvigorates antitumor immunity in mice and cancer patients, PD-1-deficient CD8 T cells exhibit a terminal exhaustion phenotype in LCMV-infected mice at later stages, despite exhibiting a ~6-fold higher expansion compared to WT T cells [
35]. During the early stage of virus infection, PD-1-deficient CD8 T cells display enhanced mitochondrial respiration and glycolysis, resulting in elevated cytokine production and cytolytic activity. This phenotype is associated with an increased expression of peroxisome proliferator-activated receptor gamma coactivator-1α (PGC1α), a master regulator of mitochondrial biogenesis [
36]. Although Tim-3 is a reliable marker of terminal exhausted T cells, its deficiency dampens the primary and secondary CD8 T cell responses during experimental Listeria monocytogenes infection [
37]. Nonetheless, inhibition of Tim-3 signaling using blocking antibodies inhibited tumor growth in mice. However, this observation may be attributed to the regulation of Tim-3-expressing dendritic cells and/or regulatory T cells, suggesting that CD8 T cell function was indirectly enhanced upon anti-Tim-3 therapy in mice [
38,
39,
40]. A plausible conclusion from these and other studies is that the expression of co-inhibitory receptors is the consequence, rather than the initiator, of T cell exhaustion.
Several transcription factors, including TOX, the NR4A family, Bhlhe40, FoxO1 and the NFAT family, have been identified as critical regulators of T cell exhaustion [
41,
42,
43,
44,
45].
Tox-deficient T
pex cells preferentially differentiate into CTL-like T cells rather than T
ex cells, thereby promoting enhanced tumor control. However, this excessive differentiation into effector cells causes a progressive loss of the T
pex population [
41,
42,
43]. The NR4A family, which consists of the three orphan nuclear receptors NR4A1 (Nur77), NR4A2 (Nurr), and NR4A3 (Nor1), plays a key role in T cell exhaustion. Ablation of all NR4A members prevents terminal exhaustion of CD8 T cells, resulting in the replacement of exhausted cells with effector-like T cells characterized by low PD-1 and Tim-3 expression [
44]. However, this does not promote the expansion of T
pex cells, suggesting that the NR4A family functions as a late-stage regulator in T cell exhaustion. However, the precise role of TOX and the NR4A transcription factors in the metabolic regulation of CD8 T cell exhaustion remains incompletely understood. Bhlhe40, a stress-responsive transcriptional regulator, has been shown to directly bind to mitochondrial genes and regulate tissue residency of T cells. Bhlhe40-deficient CD8 T cells exhibit a loss of electron transport gene expression, leading to the accumulation of dysfunctional mitochondria, metabolic reprogramming, and impaired tumor control [
46]. However, a recent study demonstrated that ablation of Bhlhe40 results in the accumulation of T
pex cells, accompanied by reduced expression of the terminal exhaustion markers [
47]. These findings reveal an intriguing paradox, as mitochondrial integrity is considered crucial for sustaining the precursor population. How Bhlhe40 affects mitochondrial gene expression in different T cell subsets and how additional functions of Bhlhe40 contribute to this complex phenotype warrants further investigation. Two complementary studies have recently demonstrated that the overexpression of FoxO1 enhances CD8 T cell stemness and boosts the ‘fitness’ of CAR T cells through metabolic reprogramming toward respiration, thereby augmenting antitumor immunity in a TCF1-independent manner [
48,
49,
50]. Of note, IL-15-treated CD8 T cells exhibit increased expression of FoxO1 and its target genes. Consistent with IL-15’s role in promoting mitochondrial respiration, dampening glycolysis, and maintaining stemness of CD8 T cells, overexpression of FoxO1 in CART cells significantly enhances mitochondrial respiration and reduces the expression of exhaustion-related genes [
48]. Conversely, reducing the glycolytic activity in T cells, such as through inactivation of the glycolytic master regulator HIF-1α, promotes a shift in CD8 T cells towards greater reliance on mitochondrial respiration, thereby enhancing their stemness during chronic viral infection [
18].
Therefore, unraveling the regulation of T cell exhaustion by cellular metabolism will facilitate the development of targeted therapies, enabling tailored and personalized strategies for cancer immunotherapy. This review will examine the metabolic factors within the tumor microenvironment that contribute to T cell exhaustion, with a particular focus on mitochondrial biology, TCA cycle, and the metabolism of glucose, lipids, and amino acids.
The TCA (or Krebs) cycle is the central metabolic junction that unites catabolic and anabolic pathways of the three major nutrient classes: carbohydrates, lipids, and amino acids. The TCA cycle generates NADH and FADH
2, which provide the proton gradient required for oxidative phosphorylation (OXPHOS), ultimately leading to ATP production. Additionally, non-essential amino acids, as well as purines and pyrimidines required for DNA replication, are synthesized from intermediate metabolites of the TCA cycle. Most TCA cycle intermediates also participate in crucial cellular processes within the cytoplasm or nucleus, including signal transduction, epigenetic regulation, posttranslational protein modifications, and redox homeostasis—all of which play pivotal roles in T cell differentiation, effector function and exhaustion.
4.1. Catabolism and Anabolism in TCA Cycle
The TCA cycle integrates both catabolic and anabolic biochemical reactions of nutrients and intermediary metabolites. Carbohydrates, lipids, and amino acids enter the TCA cycle in different ways. Glucose, the primary carbon fuel for most lymphocytes, is converted into pyruvate in the glycolytic pathway. Pyruvate is subsequently converted into acetyl-CoA in the mitochondrial matrix, which enters the TCA cycle as citrate after condensation with oxaloacetate. Fatty acids are transported into the mitochondria through the carnitine palmitoyltransferase I (CPT1A) and undergo β-oxidation, breaking down long-chain lipids into acetyl-CoA units. Amino acids enter the TCA cycle at multiple entry points, depending on their source and functional groups [
59]. Glutamine, histidine, arginine, and proline, for instance, can enter the TCA cycle as glutamate and be converted into α-ketoglutarate (α-KG). Other amino acids may enter the cycle as succinyl-CoA, fumarate or pyruvate. Different subsets of T cells may employ distinct anaplerotic strategies—replenishing TCA cycle intermediates—depending on nutrient availability, prevalent biochemical pathways, and cellular redox status (
i.e., the availability of reduction equivalents). Although all sources of TCA cycle substrates may be utilized for energy conversion and ATP production, different nutrient sources also influence the phenotype and function of T cells. For example, when T cells are activated
in vitro, they primarily utilize glucose as an energy source and adopt an effector-like phenotype. In contrast, cultivating lymphocytes with unsaturated fatty acids promotes a memory-like T cell differentiation [
60]. It is important to note that the metabolic conditions
in vitro differ significantly from those in tissues, which has major implications for nutrient utilization. For instance, while glutamine and acetate serve as primary carbon sources for energy production
in vivo, glucose is relatively scarce in most tissues [
61,
62].
4.2. The Direction of the TCA Cycle
The TCA cycle includes both reversible and irreversible reactions, offering multiple directional possibilities. The first irreversible step is the conversion of pyruvate into acetyl-CoA, which requires coenzyme A (CoA). Similarly, the conversion of α-KG to succinyl-CoA is also irreversible and consumes coenzyme A. Since both steps require the same substrate (CoA), they compete for it within the mitochondria [
63]. Under normoxic conditions, the TCA cycle proceeds oxidatively from pyruvate to citrate, succinate, fumarate, malate, and finally oxaloacetate. However, under hypoxic conditions, oxidative phosphorylation is impaired, leading to proton accumulation in the mitochondrial intermembrane space. As a result, the TCA cycle shifts towards a reductive direction. In this state, oxaloacetate is converted back to succinate, while α-KG is converted to succinate, generating high-energy phosphates to maintain the mitochondrial membrane potential [
64]. These reactions consume α-KG and lead to succinate accumulation. Since α-KG is a substrate for HIF-1α hydroxylation and proteasomal degradation, its depletion results in HIF-1α accumulation, promoting and/or maintaining gene expression for anaerobic glycolysis [
18,
65]. Compared to memory T cells, effector T cells exhibit a more reductive TCA cycle [
66]. A recent study discovered that mitochondrial isocitrate dehydrogenase 2 (IDH2) plays a crucial role in the reductive TCA cycle by catalyzing the conversion of glutamate-derived α-KG to citrate. In IDH2-deficient T cells, fatty acids are utilized to sustain the oxidative TCA cycle, leading to enhanced stemness, marked by increased TCF1 expression and cytokine secretion. This finding aligns with the role of fatty acid metabolism in promoting T cell memory, highlighting a critical role of TCA cycle direction and metabolism in T cell fate decisions.
4.3. Specific Functions of TCA Cycle Metabolites
Besides providing NADH and FADH
2 for ATP biosynthesis, intermediary metabolites of the TCA cycle serve additional biological functions, such as signal transduction, post-translational protein modification, epigenetic regulation, and cellular redox balance. For instance, mitochondrial citrate can be exported into the cytoplasm and converted into acetyl-CoA via the cytosolic enzyme ATP citrate lyase (ACLY). Alternatively, acyl-CoA synthetase short chain family member 2 (ACSS2) can produce acetyl-CoA from acetate in an ATP-dependent manner. Given that nucleo-cytosolic acetyl-CoA is an essential substrate for histone acetylation, directly influencing the expression of effector molecules such as interferon γ (IFN-γ) and granzyme B (Gzmb), recent studies have addressed the different biochemical pathways of acetyl-CoA provision in T cells [
62,
67]. During acute infection, ablation of ACLY restricts acetyl-CoA production and effector T cell responses, which could be largely compensated by acetate supplementation and an ACSS2-dependent acetyl-CoA production [
67]. However, under chronic antigenic conditions such as persistent viral infections and tumors, ACSS2 ablation significantly reduces the IFN-γ production in CD8 T cells, indicating a particular need for acetate in nutrient-restricted conditions [
62]. A recent study found that genetic ablation of ACSS2 in breast cancer causes a release of acetate into the tumor microenvironment, which supports antitumor immune responses of T cells [
68]. Of note, acetate in this scenario was oxidized and utilized via ACSS1 in mitochondria, but not ACSS2 in the cytosol. Acetyl-CoA was converted into malonyl-CoA for fatty acid biosynthesis by acetyl-CoA carboxylase (ACC) in tumor-infiltrating T cells, which led to an accumulation of lipid droplets. Pharmaceutical inhibition or genetic ablation of ACC1 redirected acetyl-CoA into TCA cycle, which in turn sustained mitochondrial bioenergetics of cytotoxic T cells and improved tumor control [
69]. Recent research has identified ACLY as the primary enzyme responsible for supplying nucleocytosolic acetyl-CoA from mitochondrial citrate in terminally exhausted T cells. Its interaction with histone acetyltransferase KAT2A promotes chromatin accessibility at numerous exhaustion-associated genes [
70]. In contrast, acetate-derived acetyl-CoA, generated via ACSS2, associates with the histone acetyltransferase p300 to sustain the expression of effector and stemness-associated genes. Thus, these findings demonstrate that the origin of acetyl-CoA—generated either from mitochondrial citrate or cytosolic acetate—influences the progression of T cell exhaustion, emphasizing that subcellular metabolic compartmentalization also plays a critical role in T cell biology.
The malate-aspartate shuttle exchanges metabolites between the cytosol and the mitochondrial matrix. This transporter system fulfills several key functions: (i) transferring NADH from the cytosol to the mitochondria, thus enhancing ATP production; (ii) regulating redox pressure by converting NADP
+ to NADPH; and (iii) mediating directly and indirectly the exchange of TCA cycle intermediates, such as malate, aspartate, α-KG, glutamate and oxaloacetate between the cytosol and the mitochondrial matrix. Within the mitochondria, oxaloacetate is converted into aspartate and then exported to the cytosol. In the cytosol, glutamic-oxaloacetic transaminase 1 (GOT1) catalyzes the conversion of aspartate and α-KG into oxaloacetate and glutamate. Ablation of GOT1 in CD8 T cells disrupts the production of serine from glutamate, which is crucial for their proliferation. Interestingly, the absence of GOT1 leads to the accumulation of aspartate without affecting α-KG levels, suggesting an increased utilization of α-KG. Phenotypically, CD8 T cells lacking GOT1 are more likely to differentiate into memory cells during bacterial infection but are less effective at controlling tumor growth [
65]. Since α-KG is vital for detoxifying ammonia, GOT1-deficient CD8 T cells are more vulnerable to ammonia toxicity, resulting in impaired cytokine production and increased apoptosis [
71].
The malate-aspartate shuttle transports malate into the mitochondrial matrix while α-KG is simultaneously exchanged into the cytosol. Nucleo-cytosolic α-KG is an important co-factor for several chromatin-modifying enzymes, including histone demethylases and the Ten-Eleven-Translocation (TET) family enzymes that regulate DNA demethylation [
72]. The importance of α-KG and DNA demethylation during T cell exhaustion is exemplified by the finding that CD8 T cells deficient in TET2 maintain a T
pex phenotype with reduced expression of terminal exhaustion markers like TOX, CD39, and 2B4 during chronic infection [
73,
74]. CAR T cells ablation of TET2 enhances their antitumor immunity and prolongs the survival of tumor-bearing mice. In addition to epigenetic modifications, α-KG regulates the enzymatic activity of α-KG-dependent oxygenases in the cytosol. Among those are prolyl hydroxylases (PHD), which mediate the proteasomal degradation of the glycolytic regulator HIF-1α. Limiting α-KG levels stabilizes HIF-1α protein expression, leading to upregulation of glycolysis-related genes that promote terminal differentiation and T cell exhaustion [
18,
65].
Collectively, metabolism of the TCA cycle forms a complex regulatory network that controls the production and distribution of metabolic intermediates, which orchestrate T cell differentiation and (dys-) function. Gaining a deeper understanding of the regulatory mechanisms of the TCA cycle in T cells under pathophysiological conditions may offer new insights into overcoming tumor immune evasion and enhancing antitumor immune responses.
The utilization of nutrients from different sources shapes the differentiation of T cells and dictates their effector functions. In the next chapters, we will highlight a few examples of how different carbon sources regulate T cell differentiation and exhaustion.
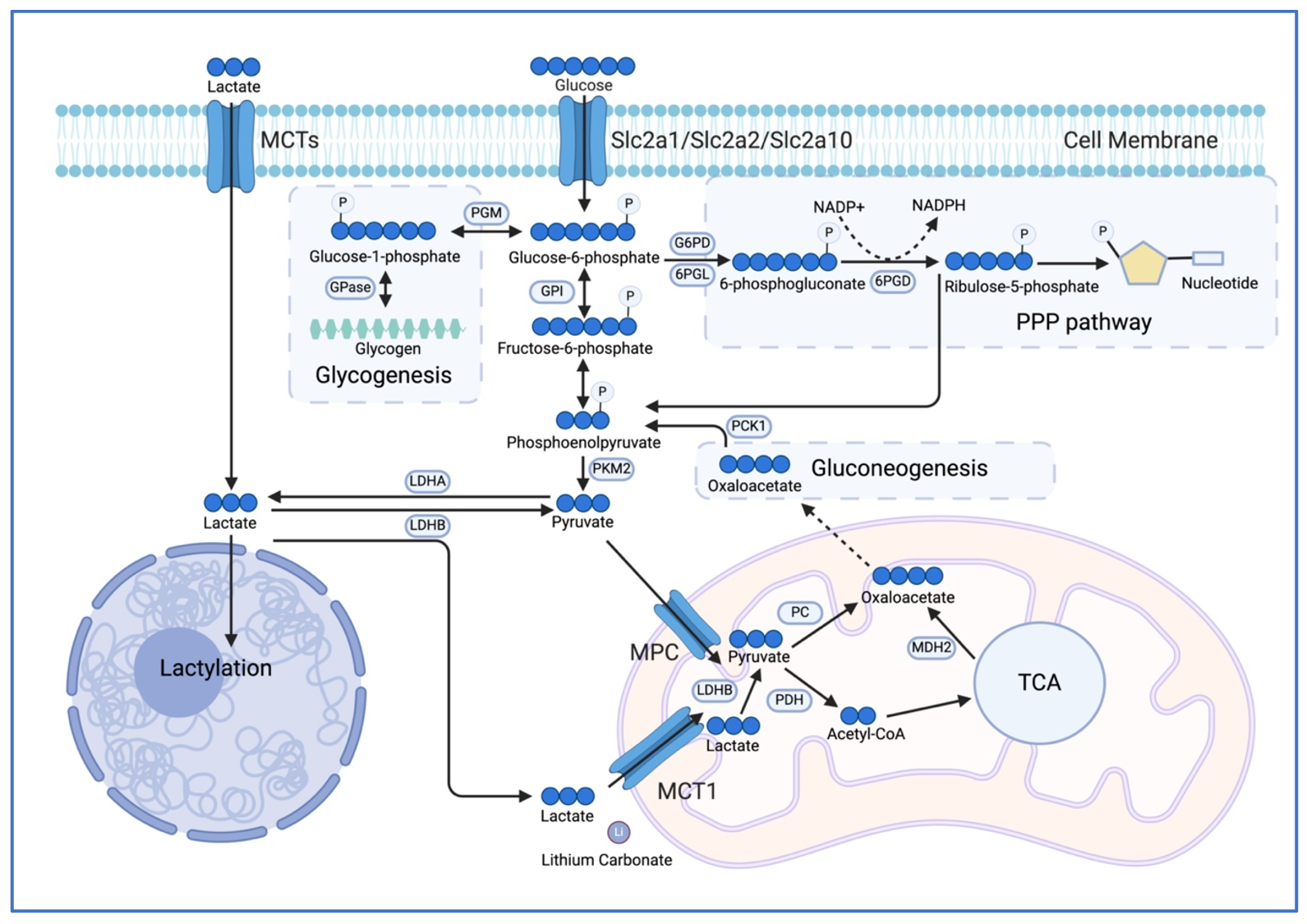
5.1. Glucose Metabolism
T cell activation upregulates both aerobic glycolysis and OXPHOS [
75]. In contrast to OXPHOS, glycolysis yields two net molecules of ATP, two molecules of NADH and two molecules of pyruvate or lactate per molecule of glucose. Nonetheless, glycolysis is the preferred metabolic pathway in activated T cells even in the presence of oxygen, a phenomenon known as the “Warburg effect”. A possible explanation for this relatively energy-inefficient metabolic process is the rapid ATP production—approximately 100-times faster than OXPHOS—and the production of essential building blocks, such as amino acids and nucleotides, which are crucial for cellular growth and rapid clonal expansion [
76]. However, recent studies revealed a complex, double-edged regulation of glucose metabolism during T cell exhaustion (). Activated and short-lived effector T cells show elevated glycolysis and glucose deprivation potently impairs T cell effector function [
77]. Early exhausted T cells also increase their glycolytic activity during chronic viral infection, whereas terminally exhausted T cells exhibited a decline in their overall metabolic activity, including glycolysis [
18]. Consistent with this, both direct or indirect restriction of glycolytic metabolism alleviates T cell exhaustion while enhancing their longevity and effector function [
78,
79].
Glucose enters T cells via glucose transporters of the SLC2A (GLUT) family. This solute carrier family contains 14 members in humans (13 in mice) and facilitates the transport of hexoses (e.g., glucose, mannose, fructose, galactose) and other structurally-related six-carbon molecules via diffusion along a concentration gradient [
80]. Several transporters were reported to be critical glucose importers in CD8 T cells including GLUT1, GLUT2, and GLUT10 but their precise role in T cell differentiation and exhaustion remains controversial [
81,
82,
83]. Intriguingly, overexpression of GLUT1 or GLUT3 augmented the metabolic fitness of CAR T cells [
84,
85,
86,
87]. GLUT1-overexpressing CAR T cells enhance not only glycolytic activity but also mitochondrial respiration, maintaining stemness and potentiating their effector function under chronic antigenic stimulation [
84,
86]. GLUT3 has a higher glucose-binding affinity than GLUT1, and it is thus assumed to be more effective at facilitating glucose uptake in glucose-limited tumor microenvironments. Ectopic GLUT3 expression improves the viability of CD8 T cells and reduces the expression of exhaustion markers, highlighting that glucose utilization also maintains the ‘fitness’ of tumor-reactive (CAR) T cells [
85,
87].
Inside T cells, glucose is rapidly phosphorylated into glucose 6-phosphate, which can be utilized in various pathways includes: (i) aerobic glycolysis, to generate pyruvate, which subsequently serves as a substrate for mitochondrial respiration; (ii) conversion of pyruvate to lactate by lactate dehydrogenase (LDHA) to regenerate NAD
+; (iii) entry into the pentose phosphate pathway (PPP) to produce NADPH and nucleotides; (iv) glycogen biosynthesis; and (v) post-translational modification, such as protein glycosylation.
Aerobic glycolysis and the conversion of pyruvate into lactate are modulated by antigen receptor signaling, which phosphorylates and activates pyruvate dehydrogenase kinase (PDHK1). Activation of PDHK1 inhibits pyruvate dehydrogenase (PDH), thereby inhibiting the production of acetyl-CoA from pyruvate and redirecting it toward lactic acid production instead. Notably, pharmacological inhibition of PDHK1 using dichloroacetate (DCA)—a clinically approved treatment for lactic acidosis—restores glucose flux into mitochondrial respiration, thereby enhancing the antitumor capacity of CD8 T cells [
88]. Moreover, a recent study demonstrated that lithium carbonate can relocate the monocarboxylate transporter 1 (MCT1), the main transporter for lactate, from the cell surface to the mitochondrial membrane. This allows T cells to utilize lactate for mitochondrial oxidation and energy production, which revitalizes exhausted T cells and potentiates cancer immunotherapy [
89].
Accumulation of lactate in the tumor microenvironment—secreted by both cancer and immune cells—dampens CD8 T cell response, partially through inhibition of NFAT activation [
90,
91]. However, ablation of LDHA in CD8 T cells severely impairs their activation, clonal expansion and effector function, primarily due to insufficient ATP production needed to sustain PI3K signaling [
76]. Besides these canonical biochemical reactions, lactate is also involved in the epigenetic regulation of CD8 T cell responses. A recent study showed that ambient lactate enhances T cell stemness and improves their antitumor immunity [
92]. In this context, lactate inhibits the activity of histone deacetylases at the TCF1 gene locus, thereby upregulating the expression of T
pex-associated genes through epigenetic rewiring. In addition to inhibiting histone deacetylation, lactate also directly alters gene expression through posttranslational lactate-modification of histone lactylation [
93]. In activated CD8 T cells, histone H3 becomes lactylated at lysine K9 and K18 [
94]. The lactate-modifications at K9 and K18 are found at both exhaustion markers (e.g., PD-1) and stemness-related genes (e.g., TCF1). Exhausted T cells showed overall lower levels of H3 lactylation compared to effector T cells. However, K9 lactylation was associated with OXPHOS genes, while K18 modifications were enriched in glycolytic genes, indicating a regulatory function of histone lactylation in different metabolic programs in exhausted T cells.
The pentose phosphate pathway (PPP) is essential for CD8 T cell function and likewise involved in T cell exhaustion. Glucose-6-phosphate dehydrogenase shunts glucose into the PPP pathway, which is metabolized in both an oxidative and non-oxidative phase to generate nicotinamide adenine dinucleotide phosphate (NADPH) and five-carbon sugars (e.g., pentose), respectively. Intriguingly, genetic or pharmacological suppression of the PPP enzyme 6-phosphogluconate dehydrogenase (6PGD) in the oxidative phase drives the differentiation of effector-like CD8 T cells with an elevated metabolic activity and better antitumor capacity [
95]. As this (short-term) effector differentiation was mediated by elevated ROS signaling, 6PGD-deficient T cells may eventually undergo terminal differentiation and functional exhaustion due to redox stress. Thus, increasing the flux through the PPP may enhance the stemness and persistence of tumor-infiltrating T cells in the long run. A case in point is the finding that pharmacological inhibition or genetic inactivation of the pyruvate kinase isozyme M2 (PKM2), which dephosphorylates phosphoenolpyruvate into pyruvate, improves the maintenance of T
pex cells in tumors. Mechanistically, suppression of PKM2 impairs the glycolytic utilization of glucose, thereby shunting glucose-6-phosphate into the PPP pathway, improving the redox resistance of T cells and enhancing checkpoint immunotherapy [
96].
Gluconeogenesis is a pathway that converts (non-) carbohydrate substrates into glucose. Pyruvate, TCA cycle intermediates and amino acids can enter glycolysis via the malate-aspartate shuttle. This process may not only be important for ‘storing’ energy and carbons in form of glycogen but also allows the redirection of metabolites into the PPP pathway to regulate redox stress when necessary. As such, Ma et al. showed that phosphoenolpyruvate carboxykinase 1 (PCK1) is required for the biosynthesis of glycogen from oxaloacetate, which is subsequently utilized in the PPP to generate abundant NADPH in stem-like CD8 T cells [
97].
However, the entire spectrum and regulation of glucose metabolism in exhausted T cells and their subsets are not yet fully understood. Integrating and adapting T cell metabolism to the metabolic microenvironment is crucial for identifying new metabolic targets to enhance antitumor immunity and for engineering CAR T cell products to better withstand the harsh conditions within tumors.
. Glucose metabolism in T cell exhaustion. Glucose metabolism, including glycolysis, pentose phosphate pathway (PPP), gluconeogenesis, and glycogenesis, plays a central role in T cell exhaustion. These pathways not only provide energy and building blocks during the T cell activation, clonal expansion, and effector differentiation, but also contribute to cellular redox regulation. Lactate is often accumulated in the tumor microenvironment and affects T cells through histone modification (lactylation), serving as a carbon resource for energy production, or inducing T cell dysfunction via acidosis and other pH-independent mechanisms. PGM: phosphoglycerate mutase; PGI: glucose-6-phosphate isomerase; G6PD: glucose-6-phosphate dehydrogenase; 6PGD: 6-phosphogluconate dehydrogenase; 6PGL: 6-phosphogluconolactonase; PC: pyruvate carboxylase; LDHA: lactate dehydrogenase-A; LDHB: lactate dehydrogenase-B; PKM2: pyruvate kinase M2; PCK1: Phosphoenolpyruvate carboxykinase 1; PDH: pyruvate dehydrogenase; MDH2: malate dehydrogenase 2; GPase: glycosyltransferase.
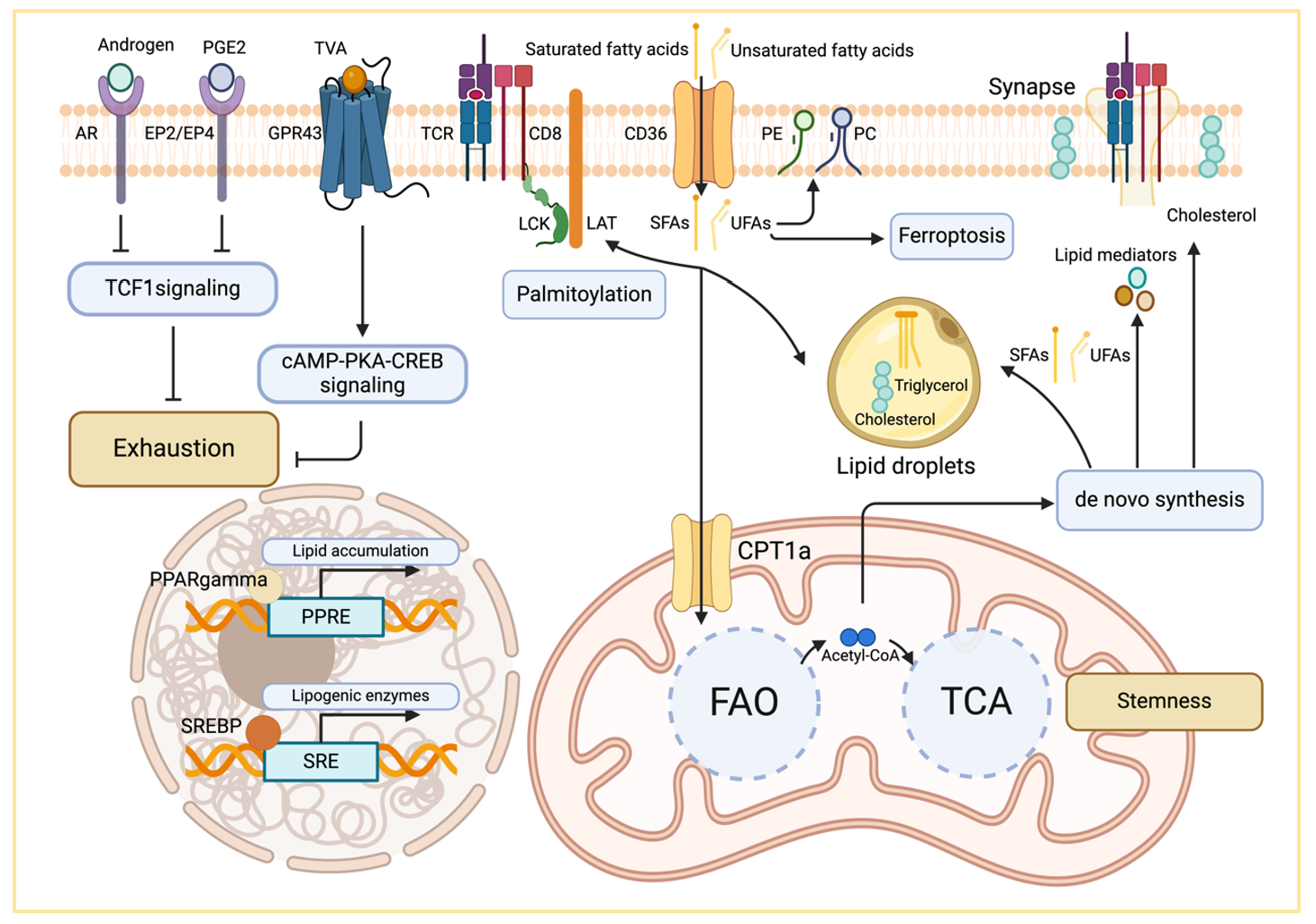
Fatty acids and lipids are not only essential components of cell membranes but also serve as nutrients, secondary messengers and redox sensors. In addition, apolar metabolites, including sterols, inflammatory mediators, and lipid hormones, regulate the differentiation, effector function, and intercellular communication of immune cells [
10,
98]. In the following section, we will briefly discuss the impact of lipid metabolism on T cell differentiation from different perspectives, including the effect of extracellular fatty acids, the utilization of intracellular lipids, and lipid-dependent epigenetic regulation ().
The tumor microenvironment is generally enriched in different lipid species [
99,
100]. Accumulated fatty acids or sterols can directly induce the dysfunction of CD8 T cells [
69,
99,
100,
101,
102]. For instance, the accumulation of long chain fatty acids in the tumor microenvironment dampens mitochondrial respiration and cytokine production in T cells [
100]. This effect could be prevented by overexpression of the very long chain acyl-CoA dehydrogenase (ACADVL) that breaks down detrimental lipids. Excess fatty acids or cholesterol are typically stored in lipid droplets—specialized intracellular organelles that not only regulate lipid metabolism but also play crucial roles in controlling lipotoxicity and inflammatory signaling. In tumor-infiltrating T cells, mitochondrial erosion impairs the capacity to break down fatty acids through β-oxidation, potentially worsening the accumulation of lipid droplets in exhausted T cells. Forcing T cells to utilize these excessive lipids by inhibition of acetyl-CoA carboxylase (ACC) potently rescued their effector function and led to improved tumor control [
103]. A high-fat diet (HFD) has been shown to significantly promote tumor growth in mouse models [
104]. In this context, tumor-infiltrating CD8 T cells exhibit both reduced numbers and impaired cytotoxic function. However, the expression of exhaustion markers—including PD-1 and Tim-3—was surprisingly unaffected or downregulated in HFD-treated tumor-bearing mice. Metabolomic analysis revealed that, in obese mice, CD8 T cells did not exhibit excessive lipid accumulation but rather displayed defects in amino acid metabolism.
It is important to note that lipids are not always a foe of T cells. A recent study showed that treatment of CD8 T cells with linoleic acid enhances their stemness
in vitro and enhances the effector function of exhausted T cells in tumors [
60]. This data corroborates that an enhancement of mitochondrial respiration is essential for CD8 T cell stemness and effector function, as discussed earlier.
. Lipid metabolism in tumor-infiltrating T cells. Fatty acids are important energy resource for the TCA cycle in T cells. Lipid-derived metabolites are also involved in signal transduction, membrane composition, and regulated cell death pathways. In addition, fatty acids serve as secondary messengers, receptor ligands, and substrates for post-translational protein modification that contribute to the differentiation and effector function of T cells in the tumor microenvironment.
5.2.1. Lipids as Membrane Components
Different fatty acids, including phosphatidylcholine (PC), phosphatidylethanolamine (PE), sphingomyelin (SM) and cholesterol, are essential components of cellular membranes. The exact composition of lipids not only controls the fluidity and stiffness of membranes but also plays important roles in transport processes, cell migration and cell division. Alterations in lipid structure and composition can trigger endoplasmic reticulum (ER) stress, senescence, or even cell death [
105,
106,
107]. Lipid peroxidation is a free radical-driven process that primarily targets unsaturated fatty acids in the plasma membrane. Peroxidation of polyunsaturated fatty acids (PUFAs) in phospholipids damages the integrity of cell membranes and induces a form of necrotic cell death called ferroptosis [
108]. CD36 is the primary lipid importer on T cells and CD36 deficiency enhances the effector function of CD8 T cells in tumor bearing mice. Mechanistically, CD36 mediated uptake of PUFAs contributes to ferroptosis of tumor-infiltrating lymphocytes, thus dampening their antitumor capacity [
109,
110]. Prevention of lipid peroxidation via Glutathione peroxidase 4 (GPX4) overexpression significantly enhances the cytokine production and the antitumor capacity of CD8 T cells [
110]. A study by Ping et al. demonstrated an important regulatory role of PC-to-PE composition in tumor-infiltrating T cells [
103]. PD-1 signaling suppresses the expression of phospholipid phosphatase 1 (PLPP1), a key enzyme involved in the synthesis of PE and PC in CD8 T cells. In the absence of PLPP1, CD8 T cells failed to metabolize unsaturated fatty acids into PE and PC, instead diverting them towards lipid peroxidation, which leads to cell death by ferroptosis. Cholesterol is also enriched within the tumor tissue and excessive cholesterol accumulation directly drives CD8 T cell exhaustion [
102]. Furthermore, membrane cholesterol plays a critical role in maintaining membrane homeostasis and fluidity [
101]. Acyl-CoA:cholesterol acyltransferase (ACAT) catalyzes the esterification of cholesterol and genetic inactivation of ACAT1 leads to an accumulation of cholesterol in the plasma membrane [
101]. Cholesterol-enriched membranes are prone to forming larger immunological synapses, resulting in an enhanced avidity of T cell antigen receptor, extended signal transduction and enhanced T cell-mediated antitumor immunity. Of note, the lipid composition of different cellular organelles differs significantly from that of the plasma membrane. The composition of the mitochondrial membrane also impacts the stemness and effector function of CD8 T cells. Cardiolipin (CL) is a mitochondrial-specific phospholipid that is localized at the inner mitochondrial membrane. The biosynthesis of CL is mediated by the protein tyrosine phosphatase mitochondrial 1 (PTPMT1) that dephosphorylates phosphatidylglycerophosphate (PGP) into phosphatidylglycerol (PG). In a second step, CL is generated by a condensation reaction between PGP and diacylglycerol (DAG). Finally, the immature (or nascent) CL is converted into its mature form by the acyltransferase Tafazzin. Ablation of PTPMT1 in T cells impairs their clonal expansion, cytokine production, and memory T cell formation [
111]. A similar phenotype was observed in mice with global Tafazzin deletion, but this defect was not observed when Tafazzin was ablated specifically in T cells. These data suggest that cardiolipin maturation is not intrinsically required for CD8 T cell function but may be important in other (immune) cell types that provide ‘help’ to cytotoxic lymphocytes. However, the investigation of lipid structures in subcellular compartments and the contribution of membrane composition to T cell differentiation, effector function and exhaustion remain in their early stages and merits further exploration.
5.2.2. Lipids as Signaling Molecules
In addition to energy metabolism and membrane components, lipids also participate in signal transduction processes that regulate T cell differentiation and exhaustion. The activation of phospholipase Cγ (PLCγ1) is an early signaling event after antigen receptor engagement. PLCγ1 hydrolyzes lipid-bound phosphatidylinositol 4,5-bisphosphate (PIP2) and generates two secondary messengers: inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG). These molecules play crucial roles in activating multiple downstream signaling pathways in T cells [
112]. DAG kinase zeta (DGKζ) converts diacylglycerol (DAG) into phosphatidic acid (PA), acting as a negative regulator of DAG-mediated signaling during T cell differentiation. In CD8 T cells, DGKζ deficiency results in pronounced T cell exhaustion in LCMV infected mice. Notably, inhibition of mTOR signaling partially rescues the phenotype of DGKζ-deficient T cells, underscoring the interplay between antigen receptor and mTOR signaling in T cell exhaustion [
113].
A recent study investigating diet-derived lipids revealed a novel role of trans-vaccenic acid (TVA) in CD8 T cell function [
114]. TVA, a long-chain unsaturated fatty acid, is not metabolized in the TCA cycle but instead regulates CD8 T cells by binding to the G-protein-coupled receptor GPR43. Inhibition of GPR43 through TVA binding counteracts the negative regulatory effects of short chain fatty acids and induces a stem-like phenotype with enhanced effector functions. Intriguingly, dietary TVA treatment was sufficient to prevent the exhaustion of tumor-infiltrating T cells, thereby leading to improved tumor control [
114]. These findings uncovered a non-canonical role of dietary fatty acids in controlling T cell immune response, and it is tempting to speculate that many more lipids may exist with potent regulatory roles.
Fatty acids are also substrates for the biosynthesis of inflammatory mediators, hormones, and cholesterol. It is worth mentioning that androgens and prostaglandins which are generated from cholesterol and arachidonic acid, respectively, play crucial regulatory roles in T cell exhaustion [
115,
116,
117,
118,
119]. Morotti et al. showed that prostaglandin E2 (PGE2) suppresses the expression of the IL-2 receptor gamma chain and mitochondrial function, thereby disrupting the expansion of T cells in the TME. Inhibition of the PGE2 receptor counteracted T cell exhaustion and restored the antitumor activity of TILs [
116]. A complementary report found that PGE2 limits the expansion and survival of T
pex cells in the TME, resulting in insufficient T cell-mediated antitumor immunity [
115]. Similar to PGE2, Androgen accelerates the exhaustion of T cells and impairs T
pex cells through epigenetic and transcriptional differentiation programs [
117]. These findings also explain the sex-bias in cancer progression and responses to cancer immunotherapy.
Lipids are also critical substrates for post-translational modifications of proteins, including palmitoylation, isoprenylation, and N-myristolation. Lipidation of proteins can enhance their hydrophobicity, thereby modulating their stability and function, including signal transduction, enzymatic activity, and conformation [
120]. Palmitoylation is one of the most common forms of lipid modification and is mediated by the palmitoyltransferase family, which contains an aspartate-histidine-histidine-cysteine (DHHC) domain responsible for catalyzing the addition of palmitoyl groups to cysteine residues. Several important signaling proteins in T cells undergo palmitoylation. Proximal signaling molecules involved in T cell activation, such as tyrosine-protein kinases and the linker for activation of T cells, are rapidly palmitoylated upon antigen receptor stimulation [
121,
122]. Moreover, PD-1 on T cells can also be palmitoylated by DHHC9 acetyltransferase, and inhibition of its lipid-based posttranslational modification has been shown to enhance the antitumor efficacy of T cells [
123]. These findings highlight the critical role of lipids in regulating signal transduction and immune checkpoint inhibition, presenting potential therapeutic avenues to improve antitumor immunity.
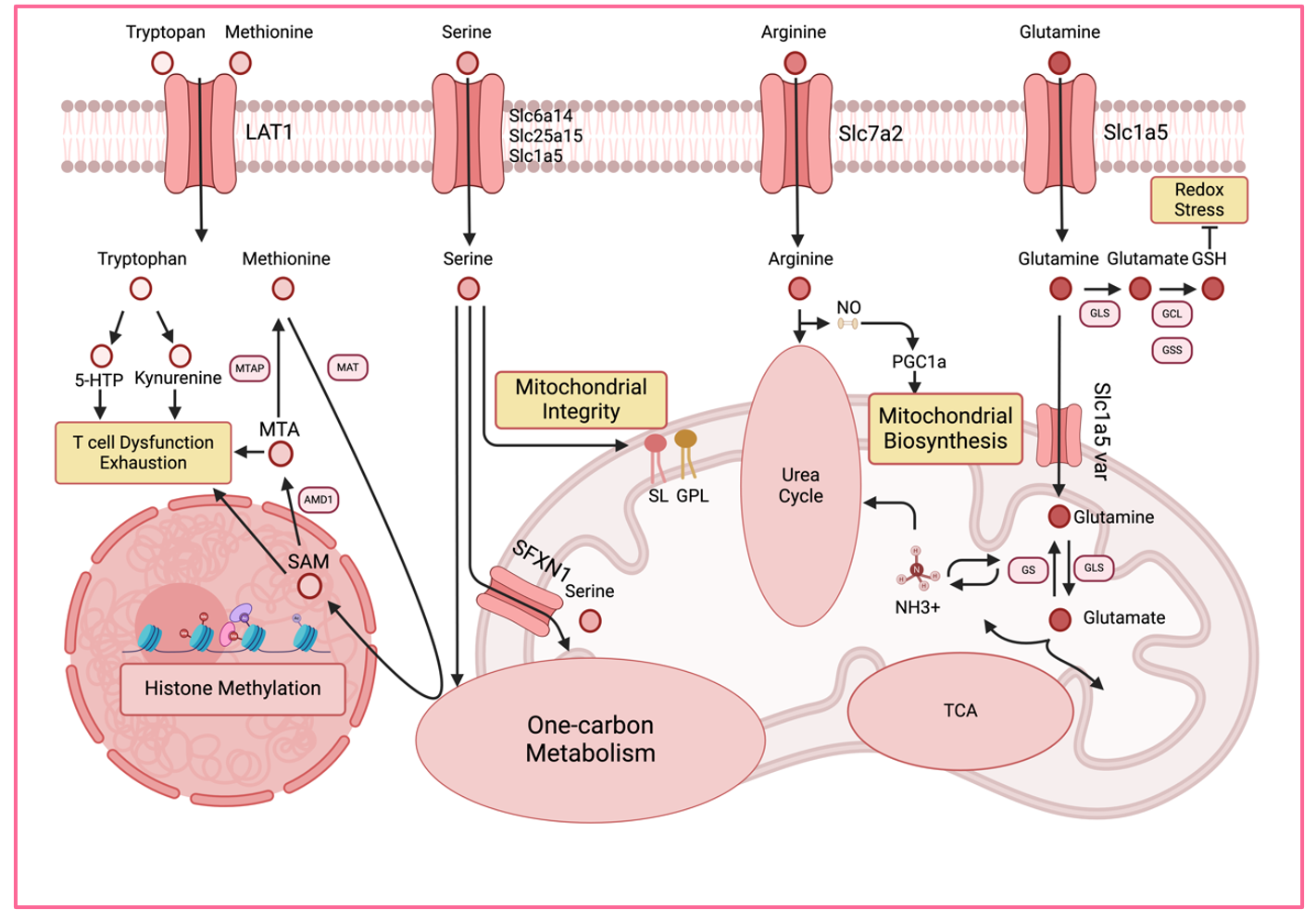
5.3. Amino Acid Metabolism
Amino acids are not only the elementary units for protein biosynthesis but also regulate gene expression, epigenetic modifications and signal transduction in T cells. The following sections will summarize a few examples of how amino acid metabolism influences T cell exhaustion ().
. Amino acid metabolism in T cell exhaustion. Amino acids play direct and indirect roles in T cell effector function and exhaustion. Tryptophan and its derivatives have diverse functions in immune cells, including the induction of T cell exhaustion via AHR signaling. The methionine cycle generates SAM, the primary methyl donor for DNA, RNA, and protein modifications. However, accumulation of SAM and MTA in the tumor microenvironment can also promote the exhaustion of T cells. Serine is a critical metabolite for effector T cells, supporting one-carbon metabolism, glutathione biosynthesis, and sphingolipid biosynthesis, which is essential for the metabolic fitness of T cells. Arginine contributes to NO production, ammonia detoxification via the urea cycle, and protein methylation, all of which impact immune regulation. Glutamine fuels the TCA cycle while also playing pivotal roles in cellular redox balance and ammonia detoxification. AHR: aryl hydrocarbon receptor; GLS: glutaminase; GCL: glutamate-cysteine ligase; GSS: glutathione synthase; GS: glutamine synthase; MAT: S-adenosylmethionine synthase; AMD1: adenosylmethionine decarboxylase; MTAP: S-methyl-5’-thioadenosine phosphorylase; NO: nitric oxide.
5.3.1. Glutamine
Glutamine is one of the most abundant amino acids in human blood, representing ~20% of total free amino acid in the serum [
124]. Glutamine plays a multifaceted role in T cell fate decision, including metabolic supplementation, epigenetic regulation, redox homeostasis, and ammonia detoxification.
The carbon skeleton of glutamine can be integrated into the TCA cycle and used for catabolic and anabolic biochemical reactions, such as gluconeogenesis and fatty acid synthesis, while its nitrogen is used for the synthesis of purines and pyrimidines. This dual role of glutamine makes it a crucial amino acid for both energy production and the biosynthesis of building blocks. Stable isotope tracing of various nutrients, including glucose, glutamine, and acetate, has demonstrated that glutamine serves as a crucial fuel for T cells
in vivo [
61]. Glutamine deprivation or glutaminase inhibition impairs the biosynthesis of polyamines and nucleotides, thereby compromising the clonal expansion of T cells. Glutamine is also essential for tumor growth and a blockage of glutamine metabolism has long been an attractive target in cancer research [
125]. However, pharmacological inhibition of glutamine utilization in mice also altered the phenotype and effector function of tumor-infiltrate lymphocytes. Remarkably, in this study T cells compensated for the glutamine restriction by utilizing acetate as an alternative fuel for the TCA cycle, which enhanced their stemness and cytokine production [
125]. However, other studies have found that glutaminase inhibition impairs the clonal expansion and effector differentiation of T cells in viral infection and antitumor immunity [
126]. These two examples emphasize the critical and context-dependent role of glutamine in T cell-mediated immunity.
Glutamine-derived nitrogen is critical for the synthesis of various important molecules including nucleotides, glucosamine, and other non-essential amino acids (NEAAs). Alanine, aspartate, proline, and serine can be synthesized from glutamine, which are then converted into asparagine, arginine, cysteine, and glycine. Notably, asparagine has been shown to rescue tumor cell survival in glutamine-deprived environments but if similar compensatory mechanisms exist in T cells remains unclear [
127].
Glutaminase (GLS) catalyzes the conversion of intracellular glutamine into glutamate. Glutamate is an important substrate for several essential metabolic pathways in T cells: together with cysteine and glycine, glutamate-cysteine ligase (GCLC) and glutathione synthetase (GS) generate glutathione, which is a potent antioxidant that is required for cellular redox regulation. Glutamate can also be converted into α-KG, which serves as an intermediate for epigenetic regulation and a TCA cycle intermediate, as discussed above.
Glutathione is critical for redox homeostasis in CD8 T cells and elevated redox stress can induce T cell exhaustion in a cell-intrinsic manner [
15,
16,
17,
18]. Indeed, conditional ablation of GCLC in T cells causes elevated ROS production, resulting in far-reaching metabolic alterations, decreased cytokine production, and impaired viability of virus-specific T cells [
128]. However, if deficiency in GCLC also causes spontaneous T cell exhaustion, remains to be demonstrated. GPX4 catalyzes the oxidation of glutathione into glutathione disulfide (GSSG) to prevent ferroptosis. Ablation of GPX4 significantly reduces the viability and cytokine production of CD8 T cells in tumors [
129]. Conversely, overexpression of GCLC or GPX4 in CD8 T cells enhances their antitumor capacity, confirming that equipping (CAR) T cells with enhanced redox resistance is beneficial for improving cancer immunotherapy.
The transamination reaction between glutamine and glutamate is critical for regulating and limiting the cellular ammonia pool. Accumulation of ammonia causes lysosomal and mitochondrial damage, resulting in defective OXPHOS, impaired effector function, and cell death. In this scenario, inhibition of glutaminase prolongs the survival of CD8 T cells in both infection and tumor models [
130]. Moreover, α-KG can be used to detoxify ammonia by generating glutamate. Decreased levels of α-KG in GOT1-deficient T cells limit the neutralization of ammonia, resulting in severe T cell dysfunction [
71].
Collectively, glutamine metabolism directly controls the proliferation, effector function, and survival of CD8 T cells. Thus, optimizing glutamine utilization and metabolism in (CAR) T cells could be an attractive antitumor strategy.
5.3.2. Arginine
Arginine is a versatile amino acid that connects various metabolic pathways and serves as a precursor for nitric oxide (NO), creatine, urea, and polyamines. Here, we highlight three key metabolic pathways related to arginine metabolism in the context of T cell exhaustion: (i) the production of nitric oxide (NO) via endothelial NOS (eNOS), (ii) ammonia detoxification via the urea cycle, and (iii) epigenetic regulation through histone methylation.
eNOS converts arginine into citrulline and releases NO, which plays distinct roles depending on its source and location. Extracellular NO, produced by myeloid-derived suppressor cells (MDSCs) in the tumor microenvironment, suppresses T cell proliferation and function, contributing to tumor immune evasion [
131]. In contrast, endogenous NO generated by eNOS within tumor-infiltrating lymphocytes enhances antigen receptor signaling, upregulates IFNγ expression, and reduces IL-2 production [
132]. Furthermore, eNOS-derived NO is involved in regulating mitochondrial respiration by inducing PGC-1α, a master regulator of mitochondrial biogenesis [
133]. However, whether these processes also occur in T cells remains currently unclear. Arginine is also an essential component of the urea cycle, where the enzyme arginase catalyzes its conversion to ornithine, thereby generating urea. This pathway is crucial for detoxifying harmful ammonia in T cells, which is essential for maintaining memory T cells [
134].
Arginine is often depleted within the tumor microenvironment, contributing to immunosuppression. Supplementing T cells with arginine can reprogram their metabolism from glycolysis to OXPHOS, enhancing T cell stemness, longevity, and antitumor immunity. Increasing arginine levels within tumors, for instance, through engineered synthetic bacteria that convert ammonia into arginine, significantly improves the antitumor capacity of T cells [
135,
136]. Beyond its roles in canonical metabolic pathways, arginine also contributes to the posttranslational modification of proteins in T cells. Protein arginine methyltransferase 5 (PRMT5) is highly expressed in T cells and catalyzes the methylation of proteins, including histones. Ablation of PRMT5 promotes the terminal differentiation of effector T cells at the expense of memory precursor cells, resulting in impaired tumor control [
137]. These findings emphasize the multifaceted roles of arginine in regulating T cell metabolism, effector function, and persistence within the tumor microenvironment.
5.3.3. Methionine
Methionine is an essential amino acid for humans, and its sustained uptake is crucial for T cell activation [
138]. This is not only due to its role in protein biosynthesis but also because of its pivotal role in the methionine cycle, which provides methyl groups for numerous biochemical reactions. Methionine adenosyltransferase II alpha (MAT2A) catalyzes the conversion of methionine into S-adenosyl-methionine (SAM), the primary methyl group donor for DNA, RNA, and protein methylation. Additionally, methionine residues in proteins act as cellular sensors for redox stress.
Cancer cells consume methionine via the
l-amino acid transporter SLC43A2, leading to methionine depletion in the tumor microenvironment. This restriction upregulates PD-1 and other exhaustion markers by altering the histone methylation landscape of tumor-infiltrating T cells [
139]. Genetic and pharmacological inhibition of SLC43A2 in tumor cells restores histone methylation in T cells and augments their antitumor capacity [
139]. Moreover, the methionine salvage pathway in cancer cells correlates with T cell exhaustion. Tumor-derived SAM and other methionine-derived metabolites induce the functional exhaustion of CD8 T cells, and ablation of MAT2A in tumor cells is sufficient to rescue the exhaustion phenotype of TILs [
140].
Another important product of the methionine cycle is homocysteine, which is utilized in the transsulfuration pathway to generate glutathione, a key molecule for maintaining cellular redox balance. As methionine is often depleted in the tumor microenvironment, targeting methionine-dependent metabolic pathways holds great promise for modulating T cell immune responses and enhancing antitumor immunotherapy.
5.3.4. Serine
Serine, synthesized endogenously from glucose or absorbed from the extracellular environment, contributes to nucleotide synthesis, one-carbon metabolism, and cellular protection against oxidative stress through glutathione biosynthesis. Although serine is a non-essential amino acid, its extracellular uptake is a rate-limiting factor for optimal T cell expansion and effector differentiation [
141]. SLC1A5, SLC6A14, SLC25A15, and sideroflexin-1 (SFXN1) have been recognized as potential serine transporters at both the plasma membrane and intracellular organelles, but the importance in T cell remains to be shown [
142,
143,
144]. In addition, genetic suppression of phosphoglycerate dehydrogenase (PHGDH), a key enzyme in serine biosynthesis, compromises T cell expansion and antitumor immunity [
141,
145,
146].
Serine plays an indispensable role in the folate cycle, acting as a one-carbon donor that is essential for purine and pyrimidine synthesis. Serine hydroxymethyltransferases (SHMT) catalyze the conversion of serine to glycine and tetrahydrofolate (THF) to methyl-THF. The mitochondrial folate cycle, driven by SHMT2, promotes mitochondrial DNA integrity, OXPHOS, glutathione biosynthesis, and ROS production [
147]. Thus, ablation of SHMT2 severely impairs T cell survival [
148]. Additionally, serine restriction or SHMT inhibition hampers T cell proliferation and pathogen clearance in murine infection models, though this effect can be reversed by exogenous nucleotide supplementation [
141].
In the tumor microenvironment, serine levels are markedly lower compared to serum concentrations, limiting effective T cell-mediated antitumor immunity by impairing one-carbon metabolism. Notably, dietary supplementation with one-carbon intermediates, such as formate, has been shown to synergize with checkpoint immunotherapy, enhancing tumor control [
149].
Serine is also a precursor in sphingolipid and glycerophospholipid synthesis, which are crucial for mitochondrial integrity, membrane fluidity, and signal transduction [
150,
151]. However, despite the critical role of serine in T cell activation and clonal expansion, its impact on T cell stemness and exhaustion warrants further exploration.
5.3.5. Tryptophan
Tryptophan, another essential amino acid, also plays critical roles in T cell-mediated antitumor immunity. Similar to methionine, tryptophan and its derivates are taken up by system L-amino acid transporters [
152]. Tryptophan is crucial for several metabolic pathways. Indoleamine 2,3-dioxygenase (IDO1/2) and tryptophan 2,3-dioxygenase (TDO) catalyze the degradation of tryptophan into kynurenine and other metabolites, including 3-hydroxykynurenine, quinolinic acid, and NAD. In addition, tryptophan hydroxylases (TPH1/2) are involved in the production of 5-hydroxytryptophan (5-HTP), serotonin, and melatonin. Although serotonin is classically known as a neurotransmitter, it is also produced by immune cells and may influence T cell activation and differentiation [
153].
IDO1/2 and TDO activity are elevated in many tumor cells and MDSCs, leading to tryptophan depletion and the accumulation of kynurenine in the tumor microenvironment [
154,
155,
156]. Kynurenine and related metabolites, such as 3-hydroxyanthranilic acid, inhibit CD8 T cell proliferation and induce apoptosis, thereby promoting tumor evasion [
157,
158]. Kynurenine also activates the aryl hydrocarbon receptor, which upregulates PD-1 and other exhaustion-associated markers [
159]. High IDO1 expression in tumors and increased levels of kynurenine and 5-HTP in the tumor microenvironment correlate with poor prognosis across multiple cancer types [
160]. Not surprisingly, pharmacological inhibition of IDO1 and/or TDO has emerged as a promising strategy to boost antitumor immunity [
154,
161]. This approach may also synergize with immune checkpoint therapy by reversing tryptophan depletion-induced T cell exhaustion [
162].
Continuous IL-2 signaling in tumor-infiltrating CD8 T cells induces TPH1 expression, which catalyzes the conversion of tryptophan to 5-HTP [
163]. In turn, 5-HTP promotes T cell exhaustion by activating AHR, leading to upregulation of inhibitory receptors and the suppression of cytokine expression [
163]. Thus, tryptophan metabolism is a potent regulator of T cell exhaustion and tumor immune evasion. Strategies aimed at restoring tryptophan availability in the tumor microenvironment or inhibiting the immunosuppressive effects of tryptophan metabolites hold great promise for enhancing T cell-mediated antitumor immunity and improving checkpoint immunotherapy.
This review examines the mechanisms of T cell exhaustion through the lens of glucose, lipid, and amino acid metabolism. Despite significant progress in understanding how cellular metabolism and the (by-) products in the tumor microenvironment affect T cell differentiation and exhaustion, many questions remain unanswered. This knowledge gap stems not from the complexity of biochemical reactions and the metabolic crosstalk in dynamic biological settings but from technical limitations in investigating metabolism at single-cell resolution.
In recent years, the metabolomic research ‘toolkit’ has expanded significantly, introducing single-cell metabolic profiling techniques, spatial metabolomics to map the distribution of metabolites in tissues, and systems biology approaches that integrate (single-cell) epigenetic, transcriptomic, and metabolomic datasets. These advancements will enable researchers to address critical questions, such as how metabolic competition between immune and cancer cells shapes the outcome of antitumor immune responses and how metabolites in the interstitial fluid influence signal transduction and epigenetics in immune and tumor cells.
CAR T cells have revolutionized cancer therapy, particularly in hematologic malignancies. However, the effectiveness of CAR T cell therapy within solid tumors remains limited due to challenges in sustaining their functionality and persistence within tumors. To overcome these challenges, engineering metabolically optimized CAR T cells that can withstand the hostile tumor microenvironment has emerged as a promising strategy to enhance the efficacy of adoptively transferred T cells. A deeper understanding of nutrient distribution, metabolic rewiring, and the interrelation of cellular metabolism with signal transduction, gene expression, and epigenetic remodeling will aid the development of novel metabolic strategies to enhance the effectiveness of cancer immunotherapy.
We thank Katrin Sinning for the contribution in figure preparation. Figures were created in BioRender (Agreement number: BR281PFB8J, UR27LABOMN, IY27XB9E7Y, HF281MHUL0).
Writing—Original Draft Preparation, H.W., M.V.; Writing—Review & Editing, H.W., M.C.P., M.V.
Not applicable.
Not applicable.
This work was supported by the Deutsche Forschungsgemeinschaft (DFG) SFB-TR 338 (“LETSimmun”)—project number: 452881907, SFB 1526 (“PANTAU”), project number: 454193335; SFB 1525 (“Cardio-Immune Interfaces”)—project number: 453989101; SFB 1583 (“DECIDE”)—project number: 49262049; and individual project grants VA882/2-1 and VA882/3-2 to M.V.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.