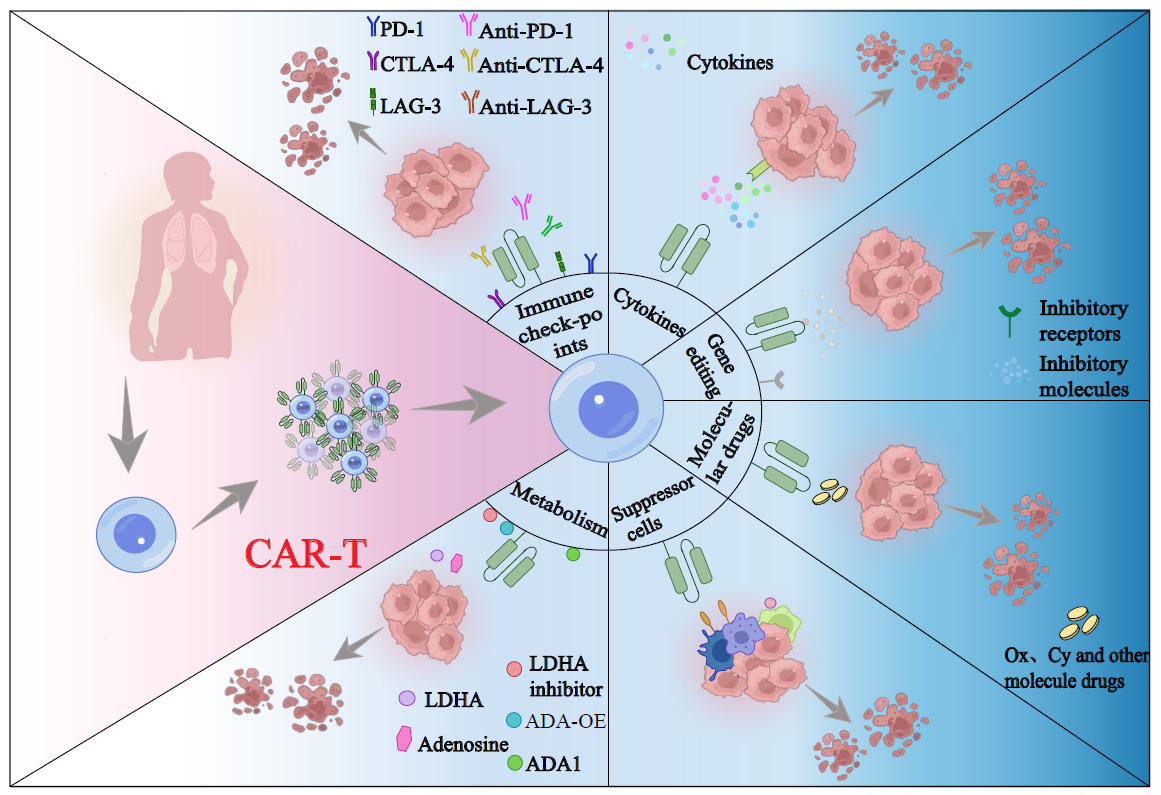
In recent years, CAR-T cell therapy, formally known as Chimeric Antigen Receptor T cell immunotherapy, has emerged as a promising form of cellular immunotherapy. This innovative treatment involves the genetic modification of the patient’s T cells ex vivo through the insertion of chimeric antigen receptors (CARs) that possess the unique ability to specifically recognize tumor-associated antigens. Following extensive expansion, these modified T cells are reintroduced into the patient’s body, thereby augmenting the immune system’s capacity to combat tumors. Notably, CAR-T cell therapy has achieved substantial outcomes in the treatment of leukemia and lymphoma patients. Some drugs targeting CD19, such as Kymriah, Yescarta, and Tecartus, have been approved by the U.S. Food and Drug Administration (FDA), marking significant milestones in this field. However, the utilization of CAR-T cells for the treatment of solid tumors continues to encounter numerous challenges.
2.1. The Brief Introduction of Structure of CAR
CAR typically consists of four structural domains: the extracellular antigen-binding domain, the hinge region, the transmembrane domain, and the intracellular signaling domain [
1,
2,
3,
4] ().
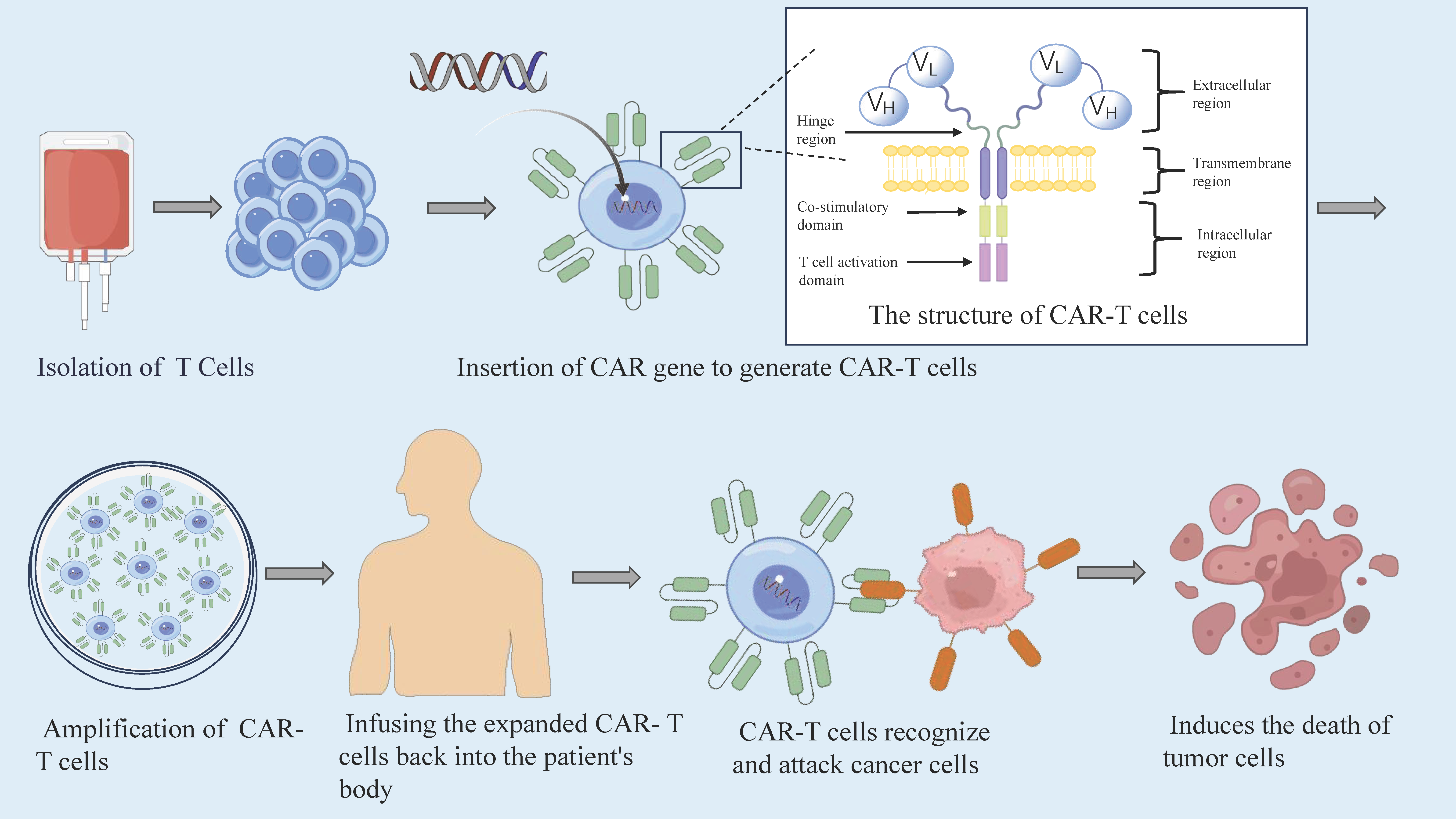
. The preparation of CAR-T cells. Extracting T cells from a patient’s peripheral blood, scientists genetically engineer these cells by inserting CAR genes to create CAR-T cells. Subsequently, these CAR-T cells are reintroduced into the patient’s body following substantial <i>in vitro</i> expansion to bolster their numbers and efficacy. This innovative approach empowers the CAR-T cells to identify and eliminate cancer cells precisely. The structure of CAR-T cells. ① The extracellular antigen-binding domain. This domain is comprised of the variable regions of an antibody’s heavy (VH) and light (VL) chains, seamlessly fused into a single-chain fragment variable (scFv) via a flexible linker. This specialized domain possesses the ability to recognize and bind with precision to epitopes presented on the surface of cells. ② The hinge region. Serving as a vital connector, the hinge region links the antigen recognition domain to the transmembrane domain. The optimal length of this region is tailored to the position of the antigen and its target epitope, thereby enhancing the CAR’s capacity for effective antigen recognition. ③ The transmembrane domain. This domain bridges the intracellular and extracellular realms, exerting a profound influence on the expression and stability of CARs. It plays a pivotal regulatory role, modulating both the expression levels of CARs and the toxicity profile of CAR-T cells. ④ The intracellular signaling domain. This region is integral to CAR-T cell function, encompassing both the co-stimulatory and signal-activating domains. The co-stimulatory domain amplifies antigen recognition signals on the CAR-T cell surface, fostering activation and proliferation. Meanwhile, the signal-activating domain is primarily responsible for transducing activation signals within CAR-T cells, with CD3ζ being the most prevalent source of activation domains in this context.
2.1.1. Antigen-Binding Domain
The antigen-binding domain of the CAR is responsible for conferring specificity to the intended target antigen (). It typically comprises the variable regions of the heavy (VH) and light (VL) chains of an antibody, joined together by a flexible linker to form a single-chain variable fragment (scFv). This scFv determines the precise specificity of antigen binding. Unlike conventional T cell receptors (TCRs) that recognize antigens presented by major histocompatibility complex (MHC) molecules, the scFv has the capability to target cell surface antigens directly. It recognizes and binds to epitopes on the cell surface, activating T cells in an MHC-independent manner and triggering an immune response [
1,
5].
2.1.2. The Hinge Region
In the evaluation of CAR functionality, the hinge region often goes unnoticed. This vital component primarily serves as a connector between the antigen recognition domain and the transmembrane domain (). Accumulating research has highlighted that the effective antigen recognition capabilities of CARs hinge on the length of the hinge region and the proximity of the target epitope to the cell membrane. Specifically, CAR-T cells are more likely to be activated when the target epitope is located close to the target cell membrane. Hudecek and colleagues conducted a study by designing three CAR structures with full-length or truncated spacers to investigate the impact of hinge domain (HD) length on CAR’s antitumor activity. Their findings revealed that while HD length did not influence CAR expression levels, CARs with medium and short spacers demonstrated superior T cell factor secretion and proliferation upon target antigen recognition [
6]. This underscores the importance of an optimal intercellular distance between CAR-T cells and target cells for the formation of the immunological synapse, which can be fine-tuned by adjusting the length of the hinge region. A hinge that is either too long or too short may impair CAR signaling [
7].
Currently, the majority of CAR hinge domains are derived from the hinge of IgG or the extracellular regions of CD8α/CD28 [
8]. IgG-based hinges typically utilize the -CH2-CH3 regions of IgG molecules, predominantly IgG
1 and IgG
4 [
9]. However, clinical studies have shown a lack of persistence in CAR-T cells featuring IgG-derived hinges [
10]. This may be attributed to specific amino acid sequences within the methionine domain binding to Fcγ receptors (FcγRs) on innate immune cells, triggering unnecessary innate immune responses, ultimately leading to the exhaustion of CAR-T cells [
11].
2.1.3. The Transmembrane Domain
The transmembrane domain (TMD) serves as the anchor for CARs on the T cell membrane, playing a fundamental role in their positioning and function (). It is commonly derived from natural proteins such as CD3ζ, CD4, CD8α, and CD28. Bridgeman and his colleagues have demonstrated that the transmembrane domain of CD3ζ facilitates T cell activation and signal transduction processes through its mediation of CAR homodimerization or interactions with endogenous TCRs [
12,
13]. By linking the proximal intracellular domain with its corresponding transmembrane domain, such as those of CD8α or CD28, one can augment the expression levels and stability of the CAR, thereby efficiently facilitating signal transduction within CAR-T cells [
5]. Collectively, the TMD plays a crucial role in influencing the stability, expression levels, dimerization, and signal transduction of CARs.
Moreover, the CAR transmembrane regions jointly influence cytokine production in CAR-T cells. Studies have demonstrated that CARs featuring an HD/TMD combination of 86 amino acids exhibit potent antitumor responses without eliciting excessive elevations in serum cytokine concentrations, thereby mitigating the risk of cytokine release syndrome (CRS) and neurotoxicity. This suggests that targeted modifications to the CAR hinge and transmembrane regions can regulate cytokine secretion and help alleviate CAR-T cell-related toxicities [
14].
Currently, the majority of TMDs in existence are derived from advancements in CAR-T therapy. In the process of designing and constructing novel CAR-T cells, it is imperative to possess sufficient and pertinent experimental data to justify the choice of the most suitable TMD [
4].
2.1.4. Intracellular Signaling Domain
In the realm of CAR engineering, the intracellular signaling domain has garnered extensive focus and research, aiming to equip CAR-modified immune cells with unparalleled antitumor immunity (). Within the CAR structure, this domain is instrumental in activating immune cells to target and attack tumor cells via efficient signal transduction. Notably, CD3ζ stands as the most prevalent activation molecule utilized in CAR-T cells.
The activation domain of CD3ζ comprises three immunoreceptor tyrosine-based activation motifs (ITAMs), designated as ITAM1, ITAM2, and ITAM3. The number and variety of ITAMs within T cells play a pivotal role in achieving optimal signaling. Intriguingly, CARs featuring a single ITAM (ITAM1, ITAM2, or ITAM3) have demonstrated superior performance compared to those incorporating triple or dual ITAMs. A study by Feucht et al. revealed that a single functional ITAM exhibits potent antitumor activity while limiting T cell differentiation, thereby promoting an increase in the proportion of central memory CAR-T cells and enhancing their persistence [
15].
The majority of CARs rely on the activation of CAR-T cells through ITAMs derived from CD3ζ [
16]. Initially, CARs harbored a single CD3ζ signaling domain [
17]. However, first-generation CARs suffered from instability and suboptimal persistence
in vitro [
18]. To augment the proliferation, persistence, and antitumor efficacy of CAR-T cells, first-generation chimeric antigen receptor (CAR) therapies have been improved by incorporating co-stimulatory domains [
19]. This involves integrating supplementary signaling molecules into CAR-T cells, thereby furnishing the secondary signal indispensable for CAR-T cell activation. This improvement facilitates an elevated recognition and eradication proficiency of CAR-T cells towards tumor cells. Notably, co-stimulatory domains exhibit distinct functionalities and metabolic characteristics; for instance, CARs fused with 4-1BB stimulate the differentiation and sustained existence of central memory T cells, fostering mitochondrial biogenesis and oxidative metabolism. Conversely, CARs paired with CD28 promote the differentiation of effector memory T cells, predominantly relying on glycolysis for their metabolic needs [
20]. In essence, the co-stimulatory domain plays a pivotal role in determining the effectiveness of CAR-T cell therapies [
1]. The integration of co-stimulatory molecules within second- and third-generation CARs further fine-tunes the functionality of CAR-T cells, exhibiting superior antitumor efficacy and augmented persistence within living organisms [
21].
2.2. Optimization of CAR Structure
2.2.1. The Evolution of CAR Designs
Previous generations of CARs, specifically the second and third generations, which incorporated CD28 and 4-1BB, respectively, have substantially improved CAR-T cell functionality. To further integrate immune checkpoint inhibitor therapy and counteract the immunosuppressive tumor microenvironment, fourth-generation CARs, also referred to as Tumor-Reactive Universal Cytokine-Mediated Killer (TRUCK T) cells, have been devised based on the foundational second-generation CARs (). These advanced CARs not only activate T cells but also secrete corresponding cytokines (such as IL-12, IL-15, and IL-18) [
22,
23,
24], thereby augmenting the infiltration of natural killer (NK) cells and macrophages and further potentiating the antitumor effect [
25,
26,
27].
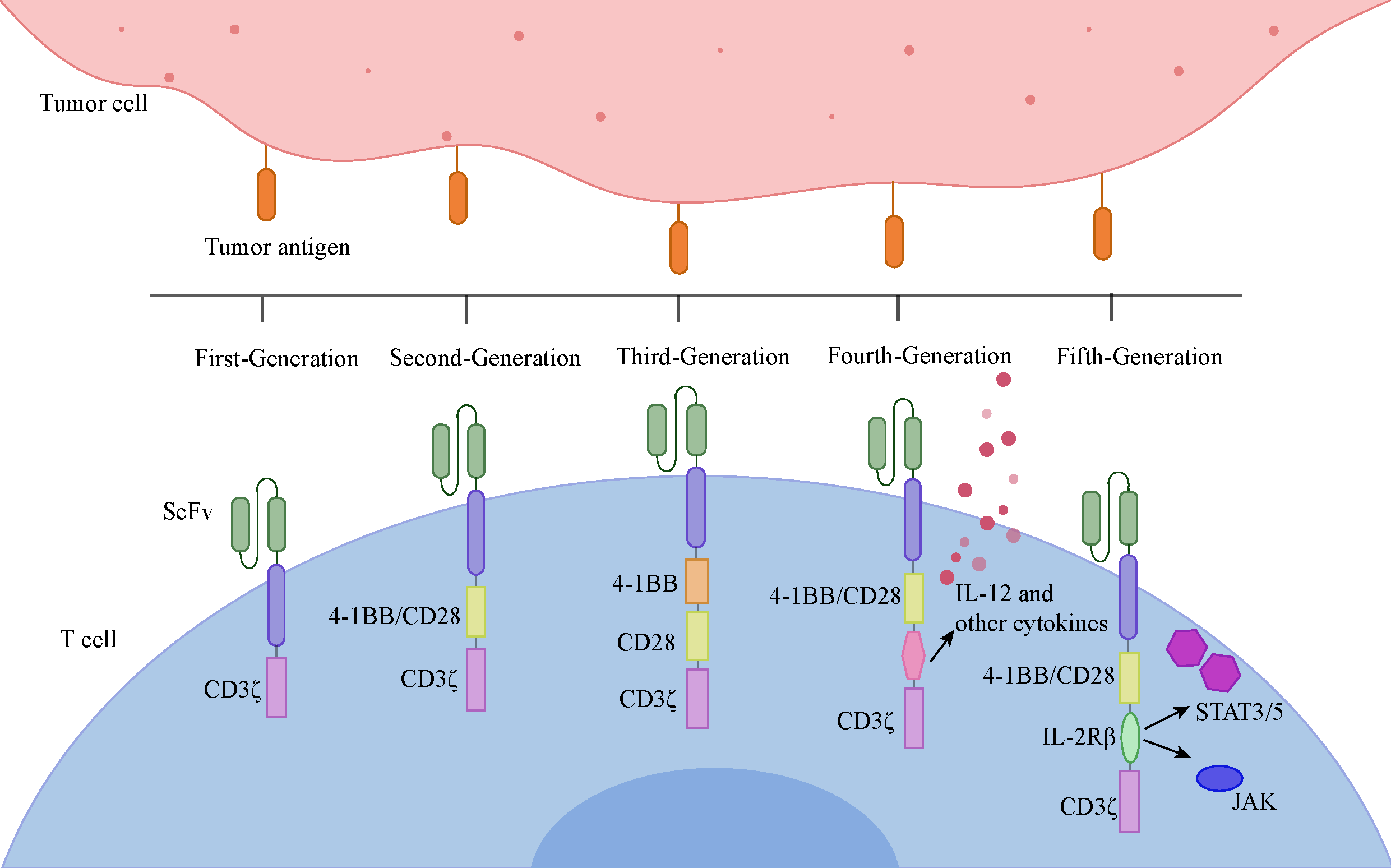
. The evolution of CAR designs. The first-generation CARs comprised solely of a single CD3ζ signaling domain. The second-generation CARs introduced an additional co-stimulatory domain on top of the first-generation design, with CD28 and 4-1BB (CD137) being the two most prevalent signaling domains utilized. The third-generation CARs featured the concurrent use of both CD28 and 4-1BB co-stimulatory domains alongside CD3ζ, though their clinical efficacy did not appear to outperform that of the second-generation CARs. Building upon the second-generation, the fourth-generation CARs incorporated the ability to express specific cytokines, such as IL-12, further amplifying their antitumor effects, they are also known as TRUCK T. The fifth-generation CARs, rooted in the second-generation CAR-T cells, integrate an intracellular domain derived from a truncated cytokine receptor (like a fragment of the IL-2Rβ chain), achieving a synergistic impact that combines antigen recognition, co-stimulation, and cytokine signaling to activate T cells comprehensively.
CAR-T cell therapy traditionally involves the genetic engineering of a patient’s T cells
ex vivo, followed by their reinfusion into the patient. However, this approach limits the scalability and translation of CAR-T cell therapy, particularly in the context of CAR-T cells. To address this limitation, researchers have developed the fifth-generation CAR, known as Universal CAR-T (). UCAR-T leverages two “third-party” systems (BBIR CAR or Super CAR) to enable CAR-T cells to recognize multiple antigens [
4,
28,
29]. The fifth-generation CAR aims to concurrently trigger the three optimal signals for T cell activation: TCR (CD3ζ domain), co-stimulation (CD28 domain), and cytokine signaling (via activation of the JAK kinase and STAT3/5 transcription factor signaling pathways). This multifaceted approach enhances CAR-T cell proliferation, survival, and antitumor activity [
30].The fifth-generation CAR’s primary advantage lies in eliminating the need to obtain T cells from patients for modification. This makes CAR-T therapy safer, and more efficient and significantly reduces treatment time and costs. However, it is important to note that such Universal CAR still poses high technical and safety challenges, and the application of the fifth-generation CAR-T cell is still in the nascent stages of exploration [
4].
2.2.2. Tandem CAR Structure
While the loss of antigen after effective CAR therapy is particularly evident in ALL patients, this phenomenon is not limited to this disease [
31,
32]. In breast cancer, bispecific CAR-T cells targeting ErbB2 and MUC1, generated
ex vivo, have shown robust antitumor activity. Engineered T cells that express a CAR specifically directed against GPC2 and concurrently secrete a bispecific innate immune cell engager (BiCE), which targets both GD2 and CD16a, exhibit enhanced intratumor retention of NK cells and augmented antitumor efficacy in cases of high-risk neuroblastoma [
33]. In a similar study, Heitzeneder et al. developed T cells expressing CARs targeting glypican-2 (GPC2), a fetal antigen expressed on neuroblastoma (NB) and several other solid tumors. Sequential GPC2-CAR design modifications significantly lowered the antigen density threshold without toxicity [
34]. Schoutrop et al. have discovered that mesothelin (MSLN)-directed CAR-T cells can effectively combat ovarian cancer by increasing CD8
+ T cell infiltration into the tumor, reducing the exhaustion of CAR-T cells, and promoting HLA-DR expression by tumor cells [
35,
36]. Pancreatic cancer is characterized by a thick stroma generated by cancer-associated fibroblast (CAF), which may contribute to the limited efficacy of MSLN-directed CAR-T cells in early-phase clinical trials. To provide a more favorable TME for CAR-T cells to target pancreatic ductal adenocarcinoma (PDAC), Wehrli et al. have generated CAR-T cells with an anti-MSLN CAR and a secreted T-cell-engaging molecule (TEAM) that targets CAFs through fibroblast activation protein (FAP) and engages T cells through CD3, termed MesoFAP CAR-TEAM cells. These cells demonstrated superior efficacy in eliminating PDAC and CAFs compared to T cells engineered to target either antigen alone [
37].
In addition, current CARs rely mainly on the activation of CAR-T cells through ITAMs derived from CD3ζ. CD3δ/ε/γ also makes an important contribution to T cell signaling. However, its potential to improve CAR performance has not been thoroughly explored. Interestingly, our team recently obtained a series of CAR-T cells against HER2 and mesothelin using a domain comprising a single ITAM from different CD3 subunits as the intracellular domain of CARs and found that one ITAM-containing CARs, especially CD3ε-derived CAR, are effective and superior to classical CARs in that they elicit a lower tonic signal, mitigated exhaustion and improved differentiation into memory populations [
38]. Similarly, another team found that CARs containing CD3δ, CD3ε or CD3γ cytoplasmic tails outperform the conventional CD3ζ CAR-T cells
in vivo [
39]. They further demonstrate that the dynamic phosphorylation of the CD3ε ITAM tyrosines is essential for CD3ε to orchestrate optimal TCR and CAR signaling and highlights the key role of CD3ε signalosome to tune signal transduction [
40]. Phosphorylation of CD3ε cytoplasmic tails by the tyrosine kinase Lck enhances ligand-induced signal initiation of both TCRs and 4-1BB-based CARs [
41]. Therefore, a better understanding of the function of the TCRs might promote rational improvement of CAR design for the treatment of cancer. These advancements illustrate the growing potential of tandem CAR designs in addressing antigen heterogeneity and enhancing the efficacy of CAR-T cell therapy across a range of malignancies.
3.1. The Intricate Tumor Microenvironment
The tumor stroma is characterized by the accumulation of various tumor-infiltrating immune cells, including tumor-associated macrophages (TAMs), regulatory T cells (Tregs), myeloid-derived suppressor cells (MDSCs), and tumor-associated fibroblasts (TAFs). This complex assembly constitutes the TME, posing significant challenges to CAR-T cell therapy (). TAMs constitute the primary immune cells within the tumor microenvironment and can be generally categorized into classically activated M1-type macrophages (M1) and alternatively activated M2-type macrophages (M2). A previous study has demonstrated that M2 macrophages facilitate the secretion of IL-8 by Tregs, which subsequently stimulates the production of transforming growth factor (TGF-β), consequently fostering an immunosuppressive microenvironment [
42]. Clinical studies have revealed that an increased infiltration of M2 correlates negatively with the effectiveness of CAR-T cell therapy and significantly impairs the proliferation of CD4
+ and CD8
+ T cells [
43]. These immunosuppressive cells produce inhibitory signals such as TGF-β, IL-10, PGE2, sFAS, Adenosine (Ado), and reactive oxygen species (ROS), which accelerate the exhaustion of CAR-T cells [
44]. Furthermore, tumor cells within the TME secrete numerous inhibitory mediators (e.g., PD-L1, TGF-β, IL-10, PGE2) by activating signaling pathways like Ras, ERK/MAPK, PI3K/Akt/mTOR. These mediators hinder the ability of T cells to effectively eliminate tumor cells and suppress the functionality of CAR-T cells [
44].
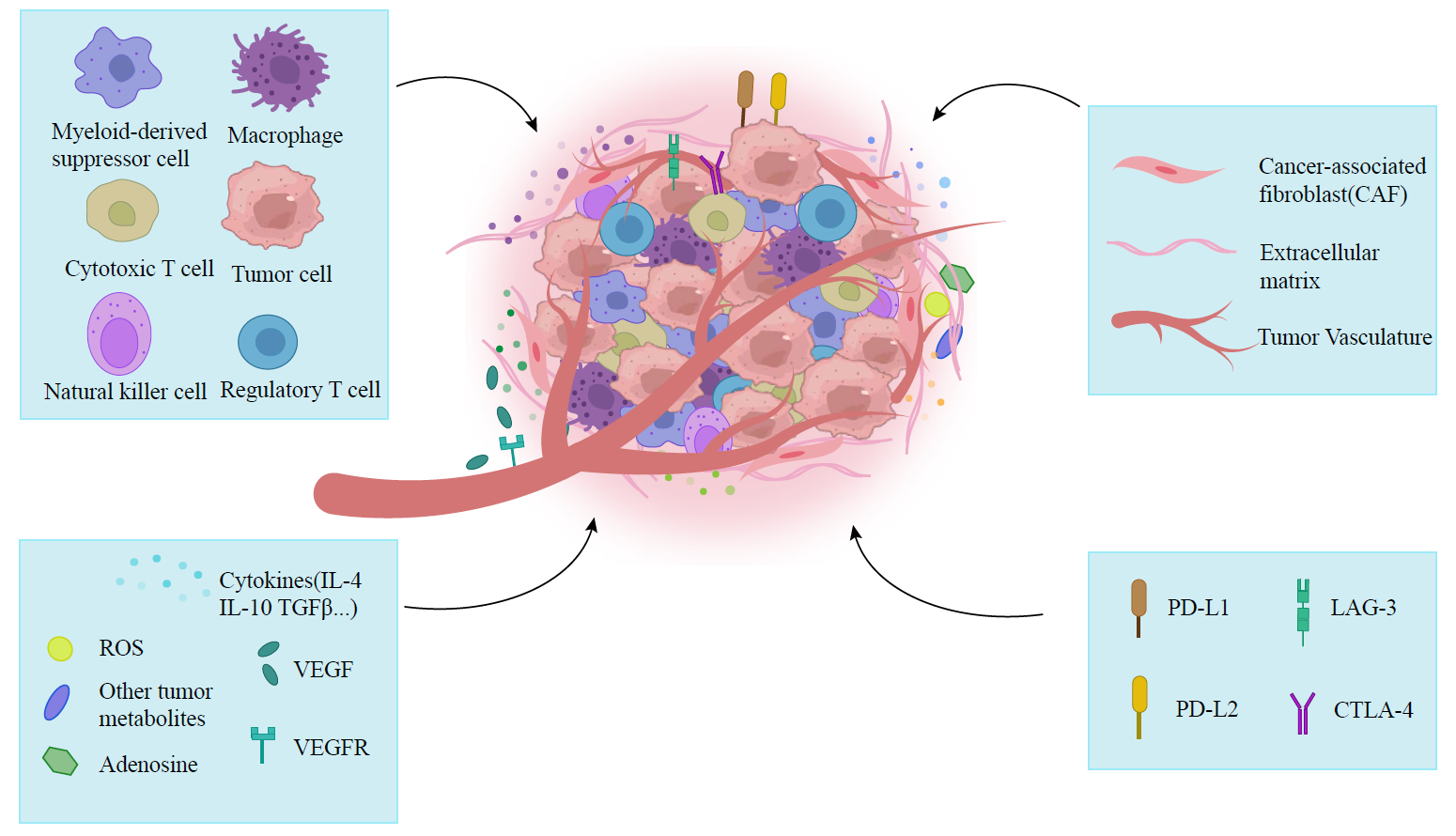
Figure 3. The complex tumor microenvironment. The complex TME is composed of a variety of components, including a multitude of cells, ECM, tumor vasculature, signaling molecules, and metabolic products of tumor cells. These structural elements, especially the ECM and tumor vasculature, create a protective barrier for tumor cells, preventing the infiltration of CAR-T cells and playing a key role in promoting tumor invasion and metastasis. Moreover, tumor cells often express inhibitory receptors or ligands, such as PD-L1, which can undermine the effectiveness of CAR-T cells. In addition to these structural and inhibitory factors, immune cells such as T cells, B cells, and NK cells can recognise tumour antigens and exert cytotoxic effects on tumor cells. Signaling molecules within the TME, including cytokines, growth factors, and chemokines, play an important role in stimulating tumor angiogenesis and recruiting immune cells. Furthermore, metabolic products generated during tumor growth, such as Ado, ROS, and lactic acid, are closely related to the growth and proliferation of tumors.
Hypoxia, another critical feature of the tumor microenvironment, promotes tumor progression through various mechanisms, including the upregulation of Ado receptor expression in immunosuppressive cells. In addition to inhibitory cells and Ado signals, the redox microenvironment and lactic acid within the TME are also significant contributors to CAR-T cell damage [
44].
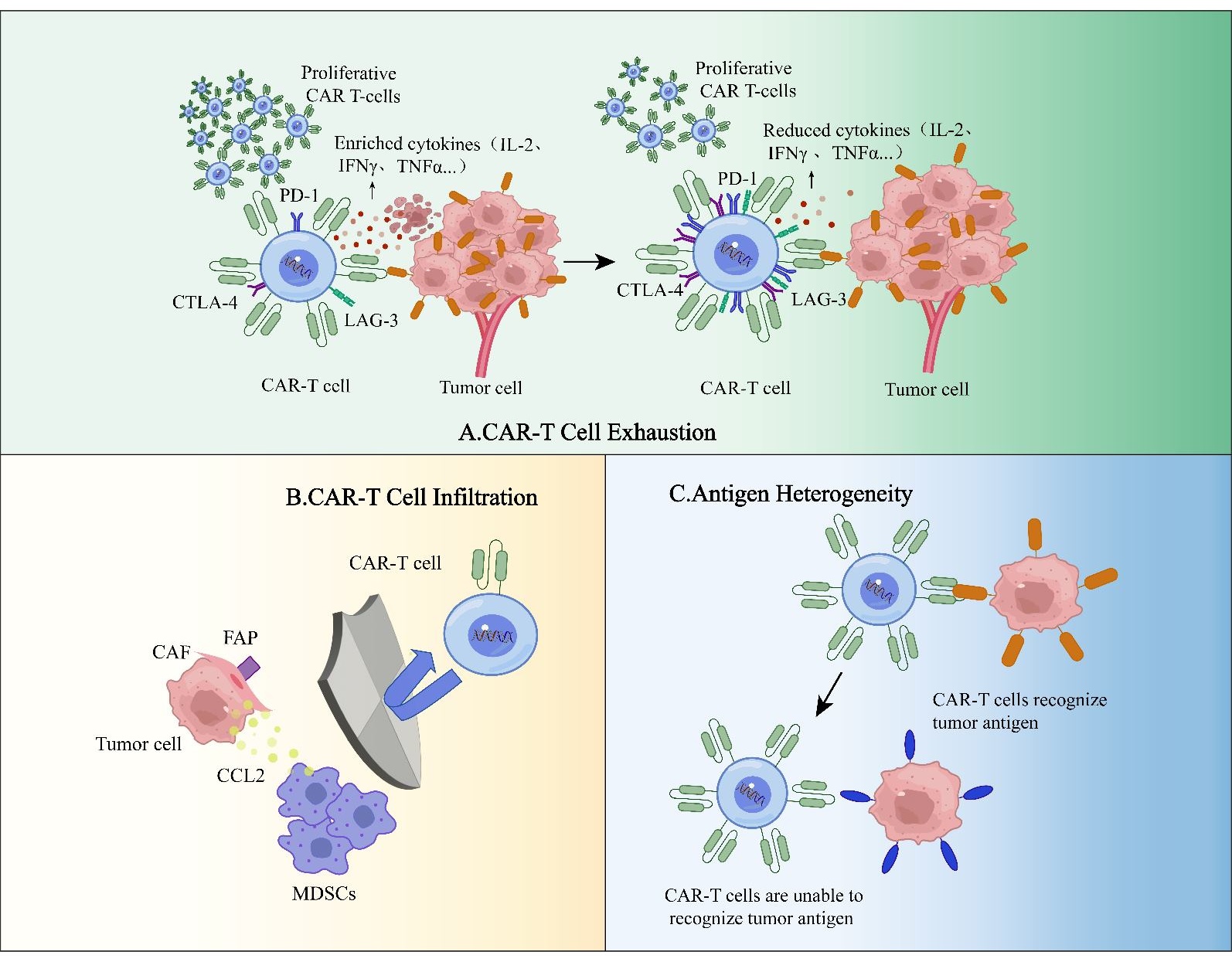
3.2. CAR-T Cell Exhaustion
Among the various factors that can impair the functionality of CAR-T cells, T cell exhaustion has emerged as a prominent and significant contributor to the limited success of CAR-T cell therapy (A). Sustained antigen stimulation is a primary driver of T cell exhaustion, where continuous exposure to disease-specific antigens leads to a decline in the proliferative capacity and effector functions of T cells, ultimately impairing their ability to recognize and eliminate tumor cells [
45,
46].
Figure 4. CAR-T cell exhaustion, infiltration and antigen heterogeneity. (<b>A</b>) Persistent antigen stimulation stands as the predominant cause of CAR-T cell exhaustion. As CAR-T cells become exhausted, they undergo a decrease in cytokine production, marked by a pronounced lack of IL-2. Furthermore, these exhausted CAR-T cells consistently demonstrate elevated expression of inhibitory receptors and reduced proliferative capacity. (<b>B</b>) CAR-T cell infiltration. The overexpression of FAP in CAFs triggers the expression of CCL2, a chemokine that enhances the recruitment of MDSCs into the TME. Consequently, this process fosters tumor growth and progression, erecting a barrier that obstructs the infiltration of CAR-T cells into the tumor tissue and impedes their penetration into the tumor site. (<b>C</b>) Antigen heterogeneity. Antigen heterogeneity can give rise to antigen escape, enabling tumor cells to evade immune attack by modifying or eliminating the specific antigens targeted by CAR-T cells, ultimately leading to decreased therapeutic effectiveness.
A notable hallmark of CAR-T cell exhaustion is a severe deficiency in the production of interleukin-2 (IL-2), a crucial factor for T cell proliferation and differentiation, which supports the growth of both effector T cells and Tregs. In the context of chronic infections and tumors, continuous antigen stimulation and inflammatory signals often propel T cells into a state of exhaustion, resulting in a substantial defect in IL-2 production [
44].
Additionally, exhausted T cells consistently exhibit heightened expression of inhibitory receptors, including programmed death-1 (PD-1), T cell immunoglobulin and mucin-domain containing-3 (TIM-3), lymphocyte-activation gene 3 (LAG-3), T cell immunoreceptor with Ig and ITIM domains (TIGIT), and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) [
44,
47,
48]. Among these, PD-1 stands out as the most prevalent inhibitory receptor associated with the induction of T cell exhaustion.
3.3. CAR-T Cell Infiltration into Tumor Sites
The inadequate infiltration of CAR-T cells into the tumor site poses a substantial challenge to the effectiveness of CAR-T cell therapy [
44] (B). Addressing this issue by enhancing CAR-T cell infiltration within the TME is critical for advancing their application in treating solid tumors. Barriers that hinder the infiltration of CAR-T cells into tumors may include: (1) The over-activation of CAFs [
49]. The overexpression of FAP in CAFs triggers their activation via the FAP-STAT3 signaling pathway, which subsequently induces the expression of CCL2. This chemokine, in turn, fosters the recruitment of MDSCs within the TME, thereby accelerating tumor growth and progression [
50]. To overcome this barrier posed by CAFs, FAP-redirected CAR-T cells can be employed to disrupt their influence, ultimately reshaping the immune microenvironment to a more favorable state for CAR-T cell infiltration and efficacy [
51,
52]. (2) The abnormal vasculature within tumors erects physical barriers that contribute to the establishment of a metabolically deranged tumor microenvironment, profoundly impeding the efficacy of CAR-T cell therapy [
44]. Vascular endothelial growth factor (VEGF) is pivotal in tumor neovascularization, facilitating angiogenesis that sustains the tumor with oxygen and nutrients, thereby playing an indispensable role in tumor progression and metastasis. (3) Furthermore, the chemokines secreted by tumor cells often fail to align with the chemokine receptors of CAR-T cells. CAR-T cells rely on their chemokine receptors (such as CXCR3 and CCR5) to recognize and bind to tumor-derived chemokines, enabling their infiltration into tumor. However, tumors frequently produce inadequate chemokines, resulting in a reduced targeting efficiency of effector cells towards the tumor. This mismatch between chemokines and their receptors undermines the effectiveness of CAR-T cell therapy [
53].
3.4. Antigen Heterogeneity in CAR-T Cell Therapy
Antigen heterogeneity describes the variability in antigen expression patterns observed within a tumor cell population. This variability can occur not only among different tumor cells but also during the progression of the same tumor cell. Such heterogeneity poses a significant threat to CAR-T cell therapy, as it can lead to antigen escape-a mechanism by which tumor cells evade immune attack by altering or losing the specific antigens targeted by CAR-T cells (C). While targeting a single antigen with CAR-T cells may initially yield a high response rate, a considerable proportion of patients treated with such therapy experience partial or complete loss of target antigen expression [
5]. This, in turn, can result in reduced therapeutic efficacy and even tumor recurrence.
Antigen escape represents a form of resistance to CAR constructs targeting a single antigen and is one of the most formidable limitations of CAR-T cell therapy. To mitigate the recurrence rate associated with CAR-T cell therapy for solid tumors, preclinical data support the adoption of strategies that target multiple antigens. An experimental investigation has shown that dual-targeting therapy aimed at IL-13Rα2 and HER2 in glioblastoma models exhibits a reduced incidence of antigen escape and a marked improvement in the capacity to eliminate solid tumor cells [
54]. By diversifying the antigenic targets, these strategies aim to overcome the challenges posed by antigen heterogeneity and enhance the durability of therapeutic responses.
Despite demonstrating promising antitumor efficacy in clinical applications, CAR-T cell therapy faces significant challenges in treating solid tumors, primarily due to the immunosuppressive nature of the tumor microenvironment and intrinsic factors related to CAR-T cells themselves. In response, scientists have been relentlessly exploring and innovating to enhance the duration of CAR activity while mitigating its exhaustion and toxicity. The CAR structure can be refined using various strategies, such as immune checkpoint inhibition, combination therapy, targeted antigen selection, gene editing, and physicochemical approaches, to enhance the anti-tumor efficacy of CAR-T cells.
4.1. Combination of Immune Checkpoint Inhibitors and CAR-T Cells
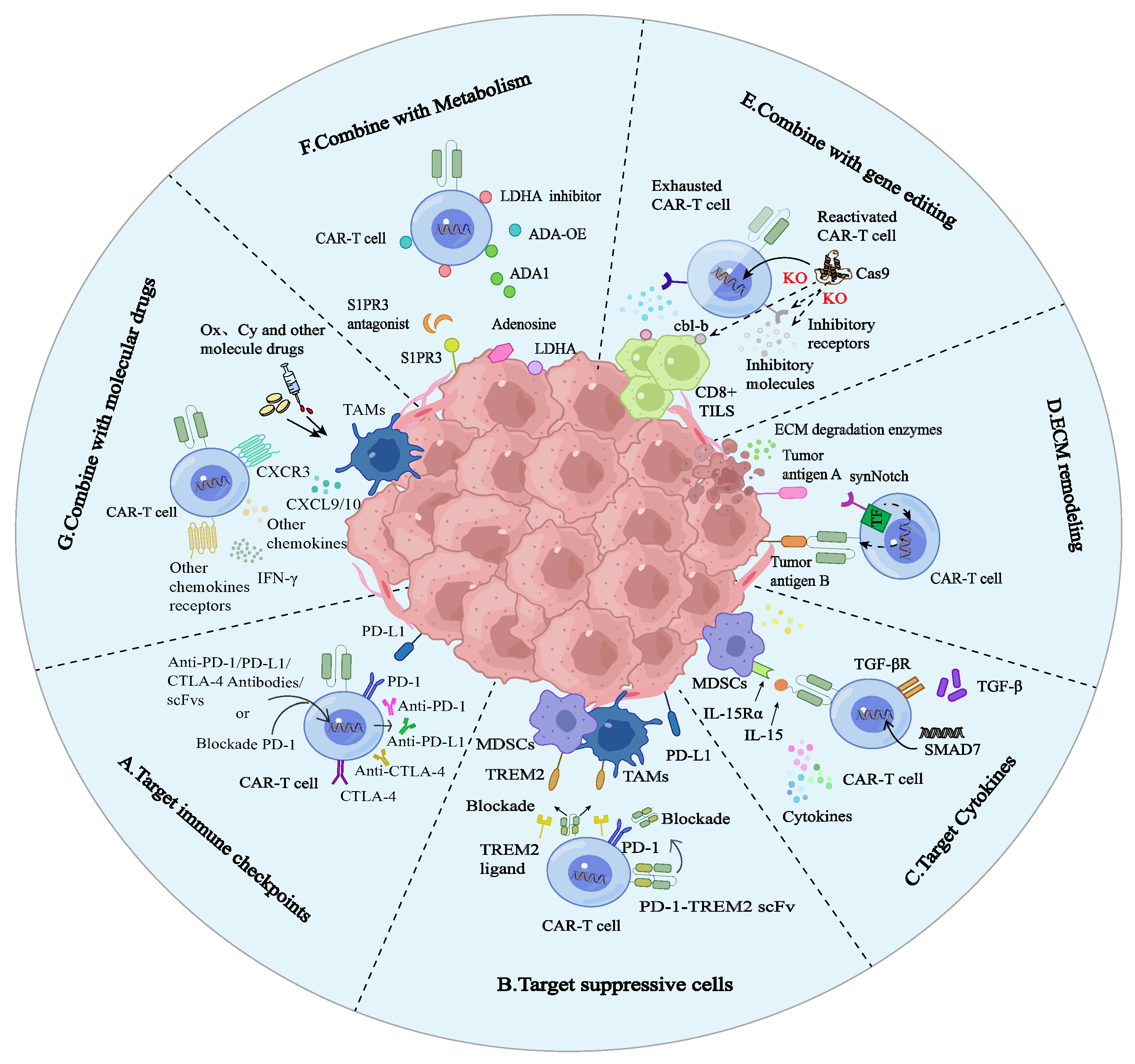
PD-1, a member of the CD28 co-stimulatory receptor family, plays a pivotal role in immune suppression by interacting with PD-L1. The PD-1/PD-L1 axis diminishes the functionality of immune cells, with persistent PD-1 expression inhibiting CAR-T cells activation and proliferation. Blockade of intrinsic PD-1 signaling has been demonstrated to effectively enhance the durability of CAR-T cells and counteract their exhaustion (A).
. The latest breakthrough in CAR-T cell therapy for solid tumors. (<b>A</b>) Targeting immune checkpoints. Employing immune checkpoint inhibitors such as anti-PD-1, anti-CTLA-4, and anti-PD-L1, or engineering CAR-T cells to secrete scFvs that target and inhibit PD-1, effectively disrupting the PD-1/PD-L1 interaction pathway. (<b>B</b>) Targeting immunosuppressive cells. By integrating an autocrine PD-1-TREM2 scFv, the PD-1/PD-L1 signaling pathway is effectively blocked, thereby impairing the functionality of MDSCs and TAMs. (<b>C</b>) Targeting cytokines. By engineering CAR-T cells to express specific cytokines, their tumor recognition and cytotoxic capabilities can be augmented. An illustrative example is the use of IL-15-modified CAR-T cells, which exhibit dual targeting capabilities against both tumor cells and MDSCs in GBM. By incorporating SMAD7 into CAR-T cells, there is a notable downregulation of inhibitory receptors, which effectively dampens the TGF-β signaling pathway and alleviates T cell exhaustion. (<b>D</b>) ECM remodeling. The synNotch receptor is capable of initiating localized secretion of ECM degrading enzymes at tumor sites, facilitating efficient tumor targeting and eradication. SynNotch CAR-T cells exhibit precise ECM recognition capabilities. In detail, upon recognizing tumor antigen A, the transcription factor (TF) module of the synNotch receptor transduces the signal into the nucleus, inducing localized CAR expression. Consequently, CAR-T cells become activated upon recognizing tumor antigen B, enabling precise elimination of tumor cells. (<b>E</b>) Gene editing. The expression of the ligase Cbl-b is upregulated in exhausted (PD-1<sup>+</sup> Tim3<sup>+</sup>) CD8<sup>+</sup> TILs. By employing CRISPR-Cas9-mediated inhibition of Cbl-b, the functions of these fatigued CD8<sup>+</sup> TILs can be rejuvenated, and CAR-T cell exhaustion can be inhibited. Furthermore, leveraging CRISPR-Cas9 to knock out inhibitory molecules and receptors contributes to restoring the functionality of exhausted CAR-T cells. (<b>F</b>) Metabolism. S1PR3, ADA-OE, cytosolic ADA1, and LDHA each contribute significantly to the regulation of the immune environment. By specifically targeting S1PR3 and manipulating the expression levels of various metabolite-associated enzymes through overexpression or inhibition, the infiltrative capacity of CAR-T cells is enhanced, thereby potentiating their antitumor efficacy. (<b>G</b>) Molecular drugs. Combination therapy involving drugs like Ox and Cy facilitates the significant release of CXCL9/10 and additional chemokines, thereby attracting immune cells and bolstering the cytotoxic activity and survival of CAR-T cells.
For cancers such as melanoma and renal cell carcinoma, the application of immune checkpoint inhibitors, including anti-PD-1, anti-CTLA-4, and anti-PD-L1, has significantly improved clinical outcomes in selected populations of patients with advanced cancer. Clinical data shows that the 5-year overall survival rate for advanced melanoma patients treated with nivolumab (anti-PD-1 antibody) in combination with ipilimumab (anti-CTLA-4 antibody) has notably risen to 52% [
55,
56,
57,
58]. A recent phase 1 trial (NCT03726515) has examined the administration of CAR-T-EGFRvIII cells in conjunction with anti-PD-1 therapy. The results indicate that CAR-T cells and PD-1 inhibition are safe and biologically effective in glioblastoma multiforme (GBM). In mouse models, Moon et al. observed that PD-1 blockade enhanced the antitumor activity of CAR-T cells targeting human melanoma [
59]. Furthermore, Suárez et al. engineered CAR-T cells to locally secrete anti-PD-L1 antibodies, promoting the infiltration of immune cell subsets into the tumor microenvironment and enhancing the efficacy of CAR-T cell therapy for solid tumors [
60]. Cherkassky et al. established that disrupting the PD-1/PD-L1 pathway through the administration of a PD-1 antibody for checkpoint blockade, the use of cell-intrinsic PD-1 shRNA for blockade, or the introduction of a PD-1 dominant negative receptor, successfully restored the effector function of CD28 CAR-T cells [
61]. Rafiq et al. engineered CAR-T cells to secrete PD-1-blocking scFvs. These modified CAR-T cells, through both paracrine and autocrine mechanisms, significantly enhanced the anti-tumor activity of both the CAR-T cells themselves and bystander tumor-specific T cells in clinically pertinent syngeneic and xenogeneic mouse models of PD-L1-positive hematologic and solid tumors [
62]. Li et al. discovered that genetically programmable cellular vesicles, engineered to express high-affinity anti-PD-L1 scFvs and loaded with a glutamine antagonist (D@aPD-L1 NVs), have been devised to metabolically disrupt the immunosuppressive TME and bolster the effectiveness of anti-MSLN CAR-T cells in orthotopic lung cancer models [
63].
The transmembrane protein CD47, an innate immune checkpoint, is crucial in preventing the immune clearance of healthy erythrocytes. By combining CAR-T cell therapy with local CD47 blockade, it is possible to reverse the immunosuppression of myelomonocytic cells, both
in vitro and within the tumor microenvironment [
64]. Another study demonstrated that EGFRvIII-SGRP CAR-T cells, constitutively secreting a signal regulatory protein gamma-related protein (SGRP) with high affinity to CD47, eradicate orthotopic EGFRvIII-mosaic GBM
in vivo, promoting glioma-associated microglia and macrophages-mediated tumor cell phagocytosis [
65].
Overall, immune checkpoint inhibitors operate by blocking inhibitory signals within the immune system, thereby enabling CAR-T cells to exert their antitumor capabilities more effectively. Clinical outcomes have demonstrated the remarkable success of these methodologies in various cancers, including melanoma, renal cell carcinoma, and lung cancer [
66].
4.2. Immunosuppressive Cell Control
The MDSCs constitute a significant proportion of the tumor stroma and typically function in innate immunity under normal conditions. However, persistent stimulation by inflammatory factors produced by tumor cells triggers the formation of MDSCs [
67]. These cells exhibit abnormal expression of immunosuppressive phenotypes, leading to a decrease in their phagocytic activity and anti-inflammatory capabilities while concurrently promoting tumor growth [
68]. Furthermore, the presence of Tregs, CD8
+ suppressive T cells, and TREM2 in the tumor microenvironment is intricately linked to tumor progression. Collectively, these factors present potential targets for CAR-T cell therapy in the treatment of solid tumors.
The presence of immunosuppressive cells significantly reduces long-term anti-tumor activity of infused CAR-T cells. Researchers are endeavoring to modify the structure of CAR-T cells by targeting specific sites, with the aim of regulating immunosuppressive cells and accomplishing an anti-tumor response (B). Research has demonstrated that blocking CXCR2 to prevent MDSC migration to tumors significantly enhances CAR-T cell infiltration and therapeutic efficacy within the tumor microenvironment [
69]. The integration of PD-L1 blockade with CAR-T cell therapy induced a phenotypic shift towards a greater proportion of M1-like macrophage subsets and the depletion of CD163
+ M2 macrophages, mediated by interferon-γ signaling, this led to an augmentation in the antitumor activity of the CAR-T cells [
70]. Chen et al. have developed CEA-specific CAR-T cells tailored to target colorectal cancer (CRC)-associated antigens, incorporating an autocrine PD-1-TREM2 scFv designed to disrupt the PD-1/PD-L1 pathway, as well as modulate MDSCs and TAMs. In a subcutaneous CRC mouse model, the PD-1-TREM2 scFv-secreting CAR-T cells exhibited remarkably potent tumor eradication capabilities, surpassing the efficacy observed with CAR-T cells secreting only the PD-1 scFv [
71]. Furthermore, Dhodapkar and colleagues have demonstrated a dynamic interplay among endogenous T cells, CAR-T cells, myeloid/dendritic cells (DCs), and tumor compartments, which significantly influences the sustained duration of the response following CAR-T cell therapy in myeloma [
72]. Rodriguez-Garcia et al. have demonstrated that CAR-T cell-mediated targeted elimination of FRβ
+ TAMs within the TME leads to an enrichment of pro-inflammatory monocytes, an influx of endogenous tumor-specific CD8
+ T cells, a delay in tumor progression, and an extension of survival [
73].
4.3. Cytokine Intervention
Cytokine engineering of CAR-T cells represents a promising approach to surmount the limited efficacy of traditional CAR-T cells against solid tumors (C). CAR-T cell mediated interferon gamma (IFNγ) production, facilitated by IL-12 signaling, is crucial for tumor cell elimination. This has been demonstrated through the engineering of an optimized membrane-bound IL-12 (mbIL12) molecule into CAR-T cells. These engineered T cells exhibit enhanced antigen-dependent proliferation, recursive tumor cell killing
in vitro, and robust
in vivo efficacy in human ovarian cancer xenograft models [
74]. Similarly, IL-12 nanostimulant-engineered CAR-T cells (INS-CAR-T) further boost the antitumor capabilities of CAR-T cells and demonstrate dramatic elimination of solid tumors [
75].
Interleukin-15 (IL-15) promotes the survival of T lymphocytes and enhances the antitumor properties of CAR-T cells in preclinical models of solid neoplasms. Patients were administered GPC3 CAR-T cells that concurrently expressed IL-15, resulting in a substantial augmentation of cell proliferation. This led to a disease control rate of 66% and an antitumor response rate of 33% [
76]. IL-15-expressing CAR-T cells exhibited reduced expression of exhaustion markers, higher antiapoptotic properties, and increased proliferative capacity upon antigen challenge. These cells promoted superior antitumor responses
in vivo in comparison with CAR-T/IL-2 cells [
77]. The co-expression of membrane-bound interleukin-15 (mIL-15) in CAR-T cells has also been shown to augment their effector functions, engraftment, tumor control, and TME reprogramming, this includes the activation of NK cells and a reduction in the presence of M2 macrophages [
78].
Interleukin-18 (IL-18) is a vital pro-inflammatory cytokine crucial for immune regulation. Lange et al. designed a novel chimeric cytokine receptor to create an autocrine loop that links activation-dependent GM-CSF production by CAR-T cells to IL-18 receptor signaling (GM18). Expression of GM18 in CAR-T cells enhanced their effector function in an antigen- and activation-dependent manner and endowed CAR-T cells with sustained effector function, resulting in potent antitumor activity in preclinical solid tumor models [
79]. Cytokine IL-18 becomes active upon caspase 1 cleavage of its latent precursor, pro-IL-18. By substituting the caspase 1 cleavage site within pro-IL-18 with a site preferred by granzyme B, a highly effective armoring strategy for γδ CAR-T cells (GzB-IL-18) is achieved, thereby enhancing their anti-tumor activity [
80]. In addition, the GD2IL-18 CAR-T cells directly against ganglioside GD2 with CAR-inducible IL-18 exhibit an enhanced response upon interacting with GD2-positive tumor cells, GD2IL-18 CAR-T cells demonstrate superior
in vivo antitumor activity, characterized by increased release of IFNγ and TNFα cytokines and more efficient target cytolysis, compared to CAR-T cells lacking inducible IL-18, resulting in the eradication of GD2-positive tumor xenografts [
81].
The production of IFNγ by CAR-T cells fosters a tumor microenvironment that is more activated and less suppressive. For example, IL-13Rα2-CAR-T cells activate patient-derived endogenous T cells and monocytes/macrophages through IFNγ signaling, inducing the generation of tumor-specific T cell responses in a GBM patient who responded positively to the treatment [
82]. A bispecific CAR targeting both IL-13Rα2 and TGF-β has also been developed, which programs tumor-specific T cells to convert TGF-β from an immunosuppressant to an immunostimulant. This approach has been shown to improve survival in both patient-derived GBM xenografts and syngeneic models of murine glioma [
83].
Interleukin-10 (IL-10) is a key anti-inflammatory cytokine that can limit immune cell activation and cytokine production. Zhao et al. have discovered that engineering CAR-T cells to secrete IL-10 promoted the proliferation and effector function of these cells, leading to the complete regression of established solid tumors and metastatic cancers across several cancer types in both syngeneic and xenograft mouse models [
84]. Furthermore, the IL-10R CAR-T cells that secret anti-CD33 bsAbs (CAR BsAb-T) exhibited an enhanced activation and induction of cytotoxicity not only in IL-10R CAR-T cells but also in bystander T cells, thereby more effectively targeting CD33-positive tumor cells [
85].
Tumor necrosis factor superfamily 14 (TNFSF14) is an interesting co-stimulatory molecule associated with T lymphocyte activation. The TNFSF14 co-expressed CAR-T cells not only showed enhanced cytotoxicity and cytokine production, but also demonstrated superior anti-tumor efficacy and improved infiltration in comparison with conventional CAR-T cells in immunodeficient NSG mice [
86].
Recent research has uncovered that inhibiting the TGF-β pathway can concurrently mitigate T cell exhaustion and curb the excessive release of cytokines (C). Consequently, the TGF-β pathway emerges as a highly promising target for optimizing CAR-T cell therapy. Our team has developed CAR-T cells that co-express SMAD7, a negative regulator of TGF-β signaling. By forming stable complexes with TGF-β receptor I (TGF-β1), SMAD7 effectively dampens the TGF-β signaling pathway. The introduction of SMAD7 into CAR-T cells led to a significant downregulation of inhibitory receptors. Notably, these modified CAR-T cells demonstrated sustained antitumor efficacy even under continuous antigen exposure. These findings validate the feasibility of targeting the TGF-β signaling pathway as a novel approach to enhance the efficacy and safety of CAR-T cell therapy for solid tumors. This research represents a significant step forward in the field, offering new hope for patients with solid tumors [
87]. Zheng et al designed T cells carrying an inverted cytokine receptor construct that fuses the TGF-β receptor II extracellular domain to the interleukin-15 (IL-15) receptor α cytoplasmic domain (named TB15). In mice with high-TGF-β solid tumors, the signal-inverted CAR/TB15 T cells effectively treat tumors by blocking TGF-β and repurposing IL-15 stimulative signaling, resulting in enhanced CAR-T cell persistence and function [
88]. Another team found that TGF-ßRIIDN-armored ROR1-CAR-T cells exhibit remarkable potency in targeting ROR1-positive tumors and exhibit resilience against the suppressive effects of TGF-β within the TME. These CAR-T cells demonstrated a significant enhancement in their restoration of cytotoxic function, along with the increased secretion of IFN-γ, TNF-α, and IL-2 in mouse xenografts of AsPC-1 pancreatic tumors [
89].
The epidermal growth factor receptor (EGFR), a member of the HER2 family, is frequently overexpressed in cancer cells and is closely associated with tumor invasion and metastasis. This characteristic has garnered significant attention from researchers in the field. Intriguingly, researchers have discovered that a mutated form of EGFR, known as EGFRvIII, is expressed in 40–70% of brain tumors. EGFRvIII features a defective extracellular ligand-binding domain, resulting in its constitutive activation in a ligand-independent manner [
90,
91]. Given the unique properties of EGFRvIII, using second-generation CARs (4-1BB/CD3ζ) specifically targeting EGFRvIII may offer a promising therapeutic approach for treating gliomas (C). By precisely targeting tumor cells that exhibit mutation-induced specific phenotypes, this “T-body therapy” not only stimulates the production of cytokines but also induces the lysis of tumor cells dependent on EGFRvIII. Preliminary studies have demonstrated the high antitumor efficacy of this CAR, positioning it as a novel and exciting target in the realm of CAR-T cell therapy [
92]. This development brings a fresh direction and renewed hope for patients suffering from gliomas. O’Rourke et al. conducted a groundbreaking first-in-human study investigating the intravenous administration of a single dose of autologous T cells equipped with a CAR specifically targeting the EGFRvIII mutation. Their findings revealed that EGFRvIII-directed CAR-T cells can mediate antigen loss and induce adaptive resistance in patients with recurrent GBM, hinting that addressing antigen heterogeneity may enhance the effectiveness of EGFRvIII-directed therapeutic strategies for GBM [
93]. CAR-T cell therapy in non-small cell lung cancer (NSCLC) is hindered by overexpression of TGF-β in the cancer cells that exert a negative regulatory role on T cell activity. EGFR-SMAD7-CAR-T demonstrated a higher proliferation rate and lysis capacity against A549 cells.
In vivo, EGFR-SMAD7-CAR-T resulted in complete tumor resorption by day 20, whereas conventional CAR-T only has a partial effect [
94]. A phase 1 study in patients shows that dual-targeting CAR-T cells (EGFR-IL-13Rα2 CAR-T cells) treatment led to target antigen reduction and cytolysis of tumor cells in glioblastoma organoids [
95]. Furthermore, an EGFRvIII CAR-T cell engineered to target acidity demonstrated high effectiveness in combating solid tumors [
96].
4.4. ECM Remodeling
The treatment of solid tumors is always hampered by the intrinsic physical microenvironment of the tumor, characterized by compact and rigid extracellular matrix (ECM) microstructures.
ECM is a unique structural component that becomes evident during the maturation and progression of tumors. As tumors expand, they exploit the process of ECM remodeling to establish a microenvironment that facilitates tumor migration, adhesion, and proliferation. Concurrently, this remodeling hinders the infiltration and cytotoxic activity of CAR-T cells, thereby suppressing the development of effective anti-tumor immune responses [
97]. Recent studies have highlighted the importance of ECM remodeling for effective cancer immunotherapy. For instance,
in-situ, oncolytic Au bioreactors have leveraged CAR-T immunotherapy to achieve a significantly enhanced inhibitory effect on tumor progression by facilitating ECM remodeling [
98].
To fully harness the potential of CAR-T therapy in treating solid tumors, it is imperative to tackle the challenges posed by the ECM. The research team led by Klabukov Ilya posits that preconditioning CAR-T cells
ex vivo could significantly boost their therapeutic effectiveness. Their hypothesis suggests that exposing CAR-T cells to specific inhibitors targeting Osr2 or HDAC3 might postpone their terminal exhaustion, ultimately enhancing the efficacy of CAR-T therapy against solid tumors. Furthermore, the direct implantation of hydrogel constructs embedded with CAR-T cells into targeted tumors could potentially aid these cells in navigating the complexities associated with the sub-endothelial basement membrane and adjacent ECM. However, these concepts require validation through rigorous modeling and clinical trials [
99]. Meanwhile, engineered CAR-T cells continue to show promise as an innovative therapeutic strategy. The synNotch CAR-T cells designed by a team exhibit precise recognition of the ECM and robust tumoricidal activity. The synNotch-induced production of enzymes enables the degradation of ECM components, thereby facilitating effective tumor targeting and elimination [
100] (D). The highly efficient and biocompatible bioorthogonal click chemistry, hyaluronidase (HAase), and the checkpoint-blocking antibody α-PD-L1 have been successfully engineered onto the surface of CAR-T cells, enhancing their therapeutic efficacy against solid tumors. The integrated HAase effectively degrades hyaluronic acid, disrupting the tumor ECM and enabling CAR-T cells to penetrate deeply into solid tumors [
101]. Furthermore, a xenografted human breast tumor model has been established to evaluate the influence of NKase, an enzyme degrading the major ECM component fibronectin, on CAR-T cell therapy. Pretreatment with NKase boosts the infiltration of CAR-T cells into tumors and thus benefits tumor inhibition [
102].
Furthermore, the characteristics of the ECM can exert an influence on tissues’ sensitivity to ionizing radiation. Research has demonstrated that adjustments to the pathways by which chemotherapeutic drugs reach tumor cells and alterations in tumors’ resistance to ionizing radiation can directly impact treatment efficacy [
103]. It is noteworthy that bioactive molecules, including peptides, proteins, and RNA deposited within the stroma, along with extracellular vesicles, present themselves as promising targets for both therapeutic interventions and diagnostic applications [
104,
105].
4.5. Gene Editing
Tumor cells employ antigen escape mechanisms to evade recognition by tumor-infiltrating CAR-T cells, posing a significant challenge in CAR-T therapy. The time required for CAR-T cells to proliferate to sufficient numbers to eliminate cancer cells can allow the disease to progress towards advanced stages. Furthermore, a single approach often cannot address all these issues comprehensively. However, advancements in site-specific gene editing and next-generation cellular engineering methods, particularly the Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)-CRISPR-associated protein 9 (Cas9) (CRISPR-Cas9) technology, offer promising solutions to these challenges (E).
Weber and his colleagues have demonstrated that the temporary cessation of antigen receptor signaling, achieved by forcing downregulation of CAR proteins or inhibiting proximal CAR signaling kinases using CRISPR-Cas9 technology, can rejuvenate exhausted T cells and restore their functionality. By engineering intermittent “rest” periods for CAR-T cells, this technology alleviates T cell exhaustion and enhances antitumor efficacy through epigenetic reprogramming [
106].
CRISPR-Cas9 editing of CAR-T cells involves targeting inhibitory genes, such as PD-1, as well as genes encoding death receptors, like CD95/Fas, to mitigate TME-mediated suppression or apoptosis. Triple knockout CAR-T cells, engineered to lack TRAC, PD-1, and B2M, have exhibited potent antitumor activity in both
in vitro and
in vivo models targeting CD19 and prostate stem cell antigen (PSCA). Building on this approach’s safety and clinical feasibility, researchers have transferred gene-edited TCR-engineered T cells into myeloma and sarcoma patients for human trials targeting the tumor-associated antigen NY-ESO-1 [
107]. MUC1 CAR-T cells, equipped with PD-1 knockout (PD-1KO), tumor-specific interleukin-12 (IL-12) release, and TGF-β receptor II knockout (TGF-βR2KO), exhibit enhanced efficacy and safety in the treatment of triple-negative breast cancer [
108]. PTG-T16R-scFV-CAR-T cells with knockdown of six inhibitory molecules, including PD-1, Tim-3, Tigit, TGF-βR, IL-10R, and IL-6R, have demonstrated greater inhibitory effects on tumor growth
in vivo and superior performance in prolonging the survival of mice. This enhancement of CAR-T cell activity against cholangiocarcinoma was achieved by simultaneous knockdown of these six inhibitory membrane proteins [
109]. The generation of PD-1-resistant T cells through CRISPR-Cas9 editing of PD-1 has been proven feasible in clinical products and shown to be safe in a Phase I trial involving patients with advanced non-small cell lung cancer [
110]. BTLA, an immune checkpoint on the surface of T cells that shares structural similarities with PD-1 and CTLA-4, inhibits CAR-T cells by recruiting tyrosine phosphatases SHP-1 and SHP-2 upon trans-engagement with HVEM. Consequently, BTLA knockout promotes CAR signaling and enhances the effector function of CAR-T cells [
111]. CD8
+ tumor-infiltrating lymphocytes (TILs) possess the unique ability to specifically recognize endogenous antigen peptide-MHC class I molecule complexes, enabling them to exert potent cytotoxic effects on tumor cells. The E3 ubiquitin ligase Cbl-b is upregulated in exhausted (PD-1
+ Tim3
+) CD8
+ TILs. Utilizing CRISPR-Cas9-mediated inhibition of Cbl-b, the effector function of these exhausted CD8
+ TILs can be restored. Depletion of Cbl-b inhibits CAR-T cell exhaustion, leading to reduced growth of MC38-CEA tumors [
112].
Employing gene editing techniques to knock down or overexpress certain key regulators of the tumor microenvironment is a strategy for enhancing CAR-T cell therapy. The nuclear factor erythroid 2-related factor 2 (Nrf2) is a key ROS-responsive factor that is implicated in increasing susceptibility to cancer progression. Knockdown of Nrf2 in human CAR-T cells has been shown to enhance the survival and function of intratumoral CAR-T cells in a solid tumor xenograft model, effectively controlling tumor growth [
113]. Good and colleagues have discovered that genetic knockdown of ID3 and SOX4 expression can improve the efficacy of CAR-T cell therapy in solid tumors by preventing or delaying CAR-T cell dysfunction. This finding suggests an NK-like CAR-T cell transition in CAR-T cell dysfunction [
114]. In another study, stable overexpression of GLUT1 in primary human CAR-T cells has been shown to improve their function and antitumor potency, with decreased T cell exhaustion and increased Th17 differentiation. This metabolic reprogramming enhances the potency of CAR-T cells against tumors [
115]. Similarly, CAR-T cells overexpressing GLUT1 or AGK specifically and effectively lysed tumor cells
in vitro in an antigen-dependent manner.
In vivo, these GLUT1- or AGK-overexpressing CAR-T cells demonstrated greater CD8
+ T cell persistence while also protecting the CAR-T cells from apoptosis upon repeated exposure to tumor cells. Furthermore, they exhibited superior antitumor efficacy in hepatocellular carcinoma (HCC) allograft mouse model [
116].
The complex TME of solid tumors poses a significant challenge to the effectiveness of CAR-T cell therapy. However, the advent of the gene editing system has paved new avenues for silencing immunosuppressive signals. By leveraging this gene-editing technology, we can explore a broader spectrum of targets and further unleash the immense potential of CAR-T cells in combating cancer.
4.6. Metabolism Regulation
An integrated strategy that merges metabolic interventions with CAR-T cell therapy promises to enhance therapeutic outcomes in the treatment of solid tumors [
117] (F).
In the current issue of Cancer Cell, Yan and colleagues elucidate how an intra-tumoral cholesterol deficiency impairs the ability of cytotoxic T cells to eliminate cancer cells by inhibiting mTORC1 signaling. Furthermore, they showcase that augmenting cholesterol levels in CAR-T cells through the inhibition of liver X receptor (LXR) results in enhanced anti-tumor functionality [
118]. Sphingosine 1-phosphate receptor 3 (S1PR3) holds significant potential in modulating the immune milieu. Utilization of an S1PR3 antagonist has been found to enhance the activation of EpCAM CAR-T cells, regulate their central memory phenotype, and mitigate CAR-T cell exhaustion
in vitro. By targeting S1PR3, the TME undergoes remodeling, which involves the recruitment of proinflammatory macrophages through the promotion of macrophage activation and polarization towards a proinflammatory phenotype. This, in turn, leads to improved infiltration of CAR-T cells and augmented recruitment of CD8
+ T cells [
119].
Adenosine (Ado), a metabolite abundantly produced within the TME, is well-known for its role in suppressing anti-tumor T cell responses through binding the Ado 2a receptor (A2aR). When A2aR expression in MSLN-CAR-T cells is targeted using the pharmacological antagonist SCH-58261, it results in the inhibition of all major anti-tumor functions of these MSLN-CAR-T cells [
120]. Exhausted CD8
+ CAR-T cells express CD39 and CD73, which catalyze the initial steps in the production of Ado. Klysz and colleagues have discovered that overexpressing Ado deaminase (ADA-OE), an enzyme that converts Ado to inosine, induces a stem-like state and enhances the functionality of CAR-T cells [
121]. In addition, a CAR-T cell was genetically engineered to express membrane-bound CD26 and cytoplasmic Ado deaminase 1 (ADA1), enabling the conversion of Ado to inosine. Upon stimulation via CD3/CD26, autocrine secretion of ADA1 occurs, which activates the CAR-T cells. This activation leads to improved migration and enhanced resistance to the suppressive effects of TGF-β1 [
122].
The excessive lactate generated through tumor glycolysis exacerbates the immunosuppression of CAR-T cells. A recent study has demonstrated that LDH-A depletion CAR-T cells showed that the dominant effect on tumor growth [
123]. The tumor microenvironment contains high lactate levels, and hypoxia enhances cellular lactate production. Hypoxia-induced signaling may influence the effectiveness of CAR-T therapy. Engineering CAR-T cells to upregulate CAR expression under hypoxic conditions induces metabolic reprogramming, reduces differentiation, and increases proliferation to enhance their antitumor activity, providing a strategy to improve efficacy and safety [
124]. Zhu et al. engineered CAR-T cells to be responsive to hypoxia by placing the CAR under the regulation of hypoxia-responsive elements. They identified the optimal structure, 5H1P-CEA CAR, which can be activated within the hypoxic tumor microenvironment. This activation leads to CAR-T cells exhibiting more durable antitumor activity [
124]. Another study presents a stringent hypoxia-sensing CAR-T cell system that effectively expresses a pan-ErbB-targeted CAR within a solid tumor, a microenvironment characterized by inadequate oxygen supply. This dynamic on/off oxygen-sensing safety switch has the potential to facilitate unlimited expansion of the CAR-T cell for treating solid malignancies [
125].
4.7. Combining Molecular Drugs with CAR-T Cells
Combining molecular targeted drugs and CAR-T cells could regulate immunity, improve TME, promote cell apoptosis, and enhance sensitivity to tumor cell killing [
126] (G). Srivastava and colleagues have discovered that a combination therapy utilizing oxaliplatin (Ox), cyclophosphamide (Cy), and an anti-PD-L1 agent exhibits a synergistic effect in enhancing CAR-T cell-mediated tumor control and survival. This finding offers a promising strategy for improving the clinical efficacy of CAR-T cells [
127]. The clinical data shows that pre-conditioning HER2-specific chimeric antigen receptor T cells (HER2 CAR-T cells) with Cy markedly transformed the immunosuppressive TME, antitumor activity was observed with clinical benefit in 50% of individuals treated [
128].
The administration of olaparib was found to substantially augment the effectiveness of EGFRvIII-targeted CAR (806-28Z CAR) T cells
in vivo. This enhancement was at least partially attributed to olaparib’s ability to inhibit the migration of MDSCs, thereby boosting the antitumor activities of CAR-T cell therapy [
129]. Treatment with the PARP inhibitor (PARPi) facilitated the infiltration of CD70 CAR-T cells by stimulating a chemokine environment conducive to CAR-T cell recruitment [
130]. The co-loading of phosphodiesterase-5 inhibitors with CAR-T cells results in the release of nitric oxide (NO), which drives vascular normalization and opens the infiltration barrier (IB), thereby enabling a greater number of T cells to enter the tumor [
131]. The administration of CAR-T cells in combination with STING agonists, such as DMXAA or cGAMP, significantly improved tumor control. This enhancement was correlated with increased CAR-T cell trafficking and persistence within the TME [
132]. Interestingly, Gardner and colleagues have devised a novel class of CAR-T cells that are genetically engineered to express an enzyme capable of activating a systemically administered small-molecule prodrug directly at the tumor site. These synthetic enzyme-armed killer cells demonstrate augmented anticancer activity when paired with small-molecule prodrugs, both
in vitro and
in vivo, in mouse tumor models [
133].
4.8. Other Methods
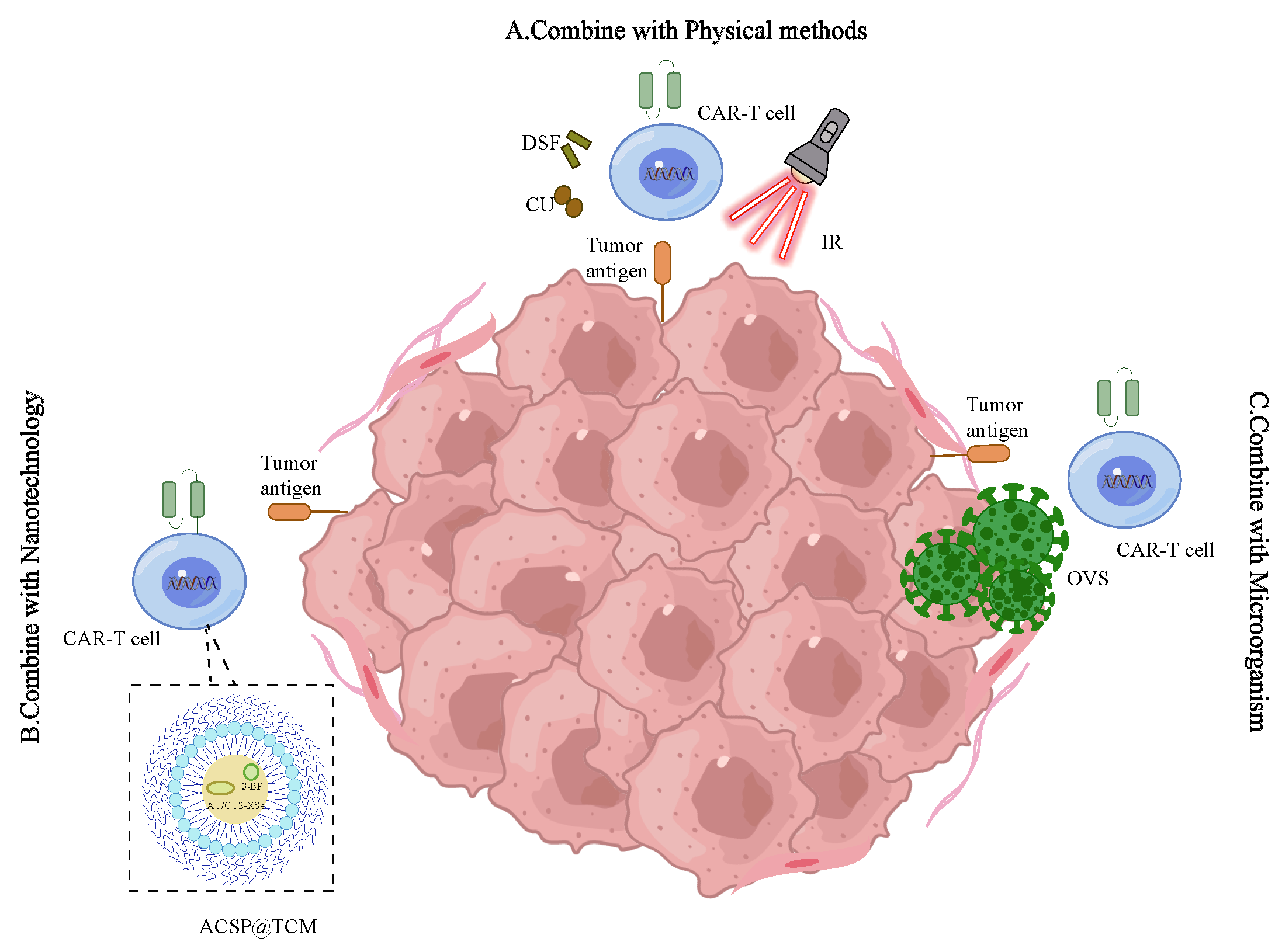
4.8.1. Physical Method
Mild hyperthermia of tumors serves to weaken their compact structure, decrease interstitial fluid pressure, augment blood perfusion, stimulate the release of antigens, and facilitate the recruitment of endogenous immune cells. Research has demonstrated that chondroitin sulfate proteoglycan-4 (CSPG4)-specific CAR-T cells exhibit superior antitumor activity following photothermal ablation of the tumor [
134]. Wang et al. have revealed that exposing CAR-T cells to the cell stress inducer disulfiram (DSF) and copper (Cu), in combination with ionizing irradiation (IR)-treated cancer cells, can endow them with early memory-like traits, robust cytotoxicity, enhanced
in vivo expansion, persistence, and reduced exhaustion [
135] (A).
. Other treatment methods. (<b>A</b>) Physical methods. By exposing CAR-T cells to the cell stress inducers DSF and Cu, and concurrently treating cancer cells with IR, we can empower CAR-T cells with robust cytotoxicity and extended efficacy, augment their proliferative abilities, and mitigate exhaustion. (<b>B</b>) Nanotechnology. A CAR-T cell membrane-camouflaged nanocatalyst (ACSP@TCM), which employs a core/shell structure encapsulating Au/Cu2-xSe and 3-bromopyruvic acid, can markedly improve the effectiveness of CAR-T cell therapy in treating solid tumors. (<b>C</b>) Microorganism. OVs, which encode a variety of transgenic elements, have been assessed as potent therapeutic agents. When integrated with CAR-T cells, OVs amplify the effectiveness of CAR-T cells within the microenvironment of solid tumors.
4.8.2. Nanotechnology
A CAR-T cell membrane-camouflaged nanocatalyst, referred to as ACSP@TCM, encapsulating a core/shell structure of Au/Cu2-xSe and 3-bromopyruvate, has been reported to significantly enhance the efficacy of CAR-T cell therapy against solid tumors [
136] (B). Additionally, nano-photosensitizer-engineered CAR-T biohybrids, known as CT-INPs, have demonstrated that mild photothermal intervention can effectively disrupt the extracellular matrix, expand blood vessels, loosen compact tissue, and stimulate chemokine secretion, all without inhibiting CAR-T cell activities. These regulatory effects create an immune-permissive tumor microenvironment conducive to the recruitment and infiltration of CT-INPs, thereby robustly amplifying the effectiveness of CAR-T immunotherapy [
137]. Integration of CAR-T therapy with nanotechnology, an injectable supramolecular hydrogel system has been designed, utilizing the self-assembly between a cationic polymer, mPEG-PCL-PEI (PPP), conjugated with a T cell-targeting anti-CD3e f(ab’)2 fragment and α-cyclodextrin (α-CD). This system is capable of loading a plasmid CAR (pCAR) with a T cell-specific CD2 promoter, successfully achieving
in situ fabrication and effective accumulation of CAR-T cells at the tumor site in humanized mouse models [
138]. Furthermore, the combination of nanozymes and B7-H3 CAR-T cells has been shown to modify the immune-hostile cancer environment, leading to enhanced activation and infiltration of B7-H3 CAR-T cells in solid tumors [
139]. Researchers have also developed a multifunctional nanocatalyst, termed APHA@CM, by encapsulating horseradish peroxidase (HRP)-loaded Au/polydopamine nanoparticles (Au/PDA NPs) and Ag2S quantum dots within CAR-T cell membranes. This nanocatalyst can precisely direct the scope and time window for nanocatalyst-induced tumor microenvironment regulation and CAR-T cell therapy in solid tumors [
140].
4.8.3. Microorganism
To address immune suppression, antigen-expressing viruses and bacteria have been engineered to elicit antitumor immune responses. The administration of live attenuated bacteria, such as Bm∆vjbR, has been shown to counteract cancer resistance to CAR-T cell therapy by modifying the TME to foster macrophage and T cell-driven antitumor immunity [
141]. Oncolytic viruses (OVs), which encode a variety of transgenes, have been evaluated as potent therapeutic agents to augment the effectiveness of CAR-modified T cells within the solid tumor microenvironment (C). In immunocompetent mouse models, Evgin et al. employed systemically delivered OVs alongside CAR-T cells to elucidate a mechanism by which OVs can enhance the efficacy of CAR-T cells against solid tumor models, including melanoma and glioma [
142]. However, it is important to note that OV infection alters the TME in multifaceted ways, some of which are beneficial, while others may be detrimental to the effectiveness of CAR-T cell therapy [
143].
4.8.4. Optimizing Culture Conditions for CAR-T Cell Manufacturing
Cytokines and stimulation conditions play a pivotal role in the production process of CAR-T cells [
144,
145]. Numerous reports have highlighted the significant impact of cytokines and growth conditions on immune T cells expansion and functional efficacy. Therefore, meticulously selecting the appropriate cytokines and refining growth conditions are indispensable for enhancing CAR-T cells proliferation and antitumor activity.
Among the various growth factors utilized in CAR-T cell cultivation, IL-7, IL-15, and IL-2 are prominent [
146]. Studies have demonstrated that IL-7 and IL-15 outperform IL-2 in sustaining CAR-T cell expansion
in vivo. Specifically, CAR-T cells cultured with IL-7 and IL-15 exhibit more sustained proliferation and higher survival rates under continuous antigen stimulation compared to those cultured with IL-2 [
147]. Consequently, their durability and antitumor activity are also superior.
In summary, refining these culture conditions can create a more conducive environment for CAR-T cells, thereby enhancing their therapeutic potential. These advancements pave the way for improved clinical immunotherapy and offer novel insights into the optimization of CAR-T cell manufacturing processes.
CAR-T therapy has progressively advanced towards clinical implementation, with more than a thousand treatments targeting diverse antigens now in clinical trials. However, the majority of these trials focus on hematological malignancies, while only a fraction, exceeding one hundred cases, address solid tumor treatments. Patients with refractory and relapsed solid tumors often face poor prognoses despite undergoing intricate multimodal therapies, necessitating the continuous development of novel therapeutic approaches.
5.1. Clinical Investigation of CAR-T Cells Targeting HER2
Human epidermal growth factor receptor 2 (HER2) is a tumor-associated protein that belongs to the EGFR family. Consequently, overexpression of HER2 is often closely associated with the occurrence and development of various cancers, such as breast cancer, ovarian cancer, and gastric cancer. Targeting HER2 has become a popular target for CAR-T cell therapy, and considerable progress has been made in multiple clinical trials of CAR-T cells targeting HER2. A Phase I/II clinical study (NCT00902044) employed HER2-CAR-T cells specifically targeting HER2 to treat 19 enrolled patients with HER2-positive tumors. Quantitative polymerase chain reaction was utilized to detect the presence of HER2-CAR-T cells. Results from nine evaluable patients revealed that the HER2-CAR-T cells persisted in the body for at least 6 weeks in seven cases. Among the 17 evaluable patients, four achieved stable disease status for durations ranging from 3 to 14 months. Furthermore, of the three patients who underwent tumor resection, one exhibited a tumor necrosis rate of ≥90%. Notably, the median overall survival for the entire cohort of 19 infused patients was 10.3 months, with a range spanning from 5.1 to 29.1 months [
148]. Furthermore, Seattle Children’s is currently conducting a Phase I clinical trial known as BrainChild-01 (NCT03500991), which is assessing the efficacy of HER2-specific CAR-T cells in treating recurrent/refractory central nervous system tumors, encompassing diffuse midline gliomas, in pediatric and young adult patients. Notably, the experimental findings have revealed high levels of CXCL10 and CCL2 in the cerebrospinal fluid of select patients, signifying robust local immune activation within the CNS. Remarkably, no toxic reactions attributable to the CAR-T cell doses were observed throughout the trial [
149]. In another Phase I clinical trial (NCT01935843), HER2-CAR-T cell infusion therapy was administered to eligible patients suffering from advanced biliary tract cancers (BTCs) and pancreatic cancers (PCs). The outcomes revealed that out of all the patients who received the infusion, one experienced a partial response that lasted for 4.5 months, while five others maintained stable disease conditions. The median progression-free survival time was 4.8 months, with a range extending from 1.5 to 8.3 months [
150].
HER2-CAR-T immunotherapy has shown promising outcomes in clinical settings, accompanied by rigorous evaluations of its safety and feasibility. These evaluations have established a solid foundation for exploratory research aimed at integrating HER2-CAR-T cell therapy with other treatments to augment the cytotoxic efficiency and persistence of CAR-T cells in targeting and eradicating cancer cells within the body.
5.2. Clinical Trials of CAR-T Cells Targeting GD2
GD2, a disialoganglioside, stands out as a proteinaceous tumor marker highly expressed in neuroblastomas and recognized as a prime therapeutic target for high-risk neuroblastomas. A recent study by Majzner et al. reported the results of a Phase I clinical trial (NCT04196413), revealing that four patients with H3K27M-mutated diffuse intrinsic pontine gliomas (DIPGs) or spinal cord diffuse midline gliomas (DMGs), who were treated with GD2 CAR-T cells, exhibited no notable tumor toxicity specifically targeted by these cells. Notably, three out of these four patients showed elevated levels of pro-inflammatory cytokines in both their plasma and cerebrospinal fluid, which coincided with clinical and radiological signs of improvement [
151]. In another Phase I single-center clinical trial (NCT02107963), 15 patients, comprising 12 with osteosarcoma and 3 with neuroblastoma, were enrolled to assess the efficacy of GD2 CAR-T cells. On the 28th day following the infusion of GD2 CAR-T cells, 16.7% of the evaluable patients showed disease progression, while a favorable 83.3% maintained stable disease (SD). However, sixty days post-infusion, only 30% of these SD patients continued to show stability, with all patients ultimately experiencing disease progression [
152]. Furthermore, the results of a Phase I clinical trial (NCT00085930) highlighted the clinical outcomes of 19 high-risk neuroblastoma patients who received GD2 CAR-T cell infusions. At the time of infusion, 8 patients were in remission, and among the 11 patients with active disease, a remarkable 3 achieved complete remission [
153].
5.3. Clinical Exploration of CAR-T Cells Targeting MSLN in Combination with Anti-PD-1 Therapy
The immune suppressive effects mediated by PD-1 are widely acknowledged as a pivotal limiting factor in the application of CAR-T cell therapy for solid tumors. Likewise, MSLN, which exhibits high expression levels across a diverse array of solid tumors but remains at low levels in normal tissues, stands out as an enticing target for cancer immunotherapy. A Phase I clinical trial, which explored the efficacy of regional MSLN-targeted CAR-T cell therapy in conjunction with the anti-PD-1 agent pembrolizumab for patients with malignant pleural disease, researchers demonstrated that PD-1 blockade exerted the anticipated therapeutic effects in mice, and subsequently, 18 patients with malignant pleural mesothelioma (MPM) also safely received pembrolizumab. The trial’s results revealed that eight patients maintained stable disease for six months, two patients demonstrated complete metabolic responses on PET scans, and the median overall survival following CAR-T cell injection was 23.9 months. These findings underscore the safety and feasibility of locally administered MSLN-targeted CAR-T cell therapy [
154]. In another study, researchers leveraged CRISPR-Cas9 technology to create PD-1 and TCR-deficient MSLN-specific CAR-T (MPTK-CAR-T) cells. Among the 15 patients who underwent one or more infusions of MPTK-CAR-T cells, no dose-limiting toxicities were reported. Two patients exhibited stable disease as a positive response, with circulating MPTK-CAR-T cells peaking between days 7 and 14 post-infusion. Notably, in three patients, TCR-positive CAR-T cells were predominantly detected in effusion fluid or peripheral blood after the infusion [
155]. These findings offer compelling data to support further investigation into the combined use of CAR-T cells and PD-1 blockers in immunotherapy for solid tumors.
5.4. Clinical Investigation of CAR-T Cells Targeting CLDN18.2
Claudin18.2 (CLDN18.2), a member of the Claudin protein family, is located on the cell membrane and typically expressed at low levels in differentiated epithelial cells of the gastric mucosa. Under pathological conditions, the expression of CLDN18.2 is significantly upregulated in various tumors, making it a promising target for cancer therapy. Satri-cel/CT041 is an autologous CAR-T cell targeting CLDN18.2.
Here, we present the Phase 1 clinical trial findings from the CT041-CG4006 study: Among the eight enrolled patients, the overall response rate amounted to 38.8%, while the disease control rate reached a commendable 91.8%. Median progression-free survival stood at 4.4 months, and median overall survival extended to 8.8 months. This pivotal trial rigorously evaluated the safety and efficacy of Satri-cel in the management of patients diagnosed with CLDN18.2-positive advanced gastrointestinal cancer (NCT03874898) [
156]. Furthermore, a comprehensive analysis that pooled data from Phase I and Ib trials examining the infusion of CT041 in patients with CLDN18.2-positive PC revealed encouraging outcomes: Thirteen patients maintained SD, translating to a disease control rate (DCR) of 70.8% (17/24). Notably, four patients exhibited partial response (PR), yielding an overall response rate (ORR) of 16.7% (4/24) (NCT03874897, NCT04581473) [
157]. Ongoing clinical trials continue to underscore the promising therapeutic efficacy and favorable safety profile of CT041. Looking ahead, CT041 holds the potential to emerge as a cornerstone treatment for gastric and pancreatic cancer, offering hope and benefit to a vast patient population worldwide.
5.5. Clinical Trials of CAR-T Cells Targeting PSMA
Prostate-specific membrane antigen (PSMA) is abundantly expressed in mCRPC, making it an ideal target for CAR-T cell therapy. This novel CAR-T cell therapy, specifically designed to target PSMA, has been further optimized by integrating the dominant-negative receptor of the immunosuppressive factor TGF-β, known as TGF-βRII (TGF-β-RDN). By blocking TGF-β signaling and addressing local immunosuppression, this therapy aims to bolster the patient’s antitumor immunity.
In a Phase I clinical trial (NCT03089203) involving mCRPC patients, the CAR-T-PSMA-TGF-β-RDN therapy exhibited encouraging results. One patient experienced a significant proliferation of CAR-T cells, accompanied by a reduction in Prostate-specific antigen (PSA) levels by more than 98%. Additionally, three patients showed a PSA reduction of ≥30%. Cell kinetics analysis revealed a notable infiltration and proliferation of CAR-T cells within the tumor tissue [
158].The CAR-T-PSMA-TGF-β-RDN therapy has demonstrated a considerable degree of safety and feasibility in clinical trials.
CAR-T cell therapy, an innovative form of cellular therapy, has demonstrated remarkable antitumor efficacy in hematological malignancies. However, its effectiveness in solid tumor treatments is significantly influenced by the complex TME. The tumor stroma poses a barrier to CAR-T cell infiltration, while immunosuppressive molecules produced by immune-inhibitory cells within the TME further impair T cell function. Additionally, prolonged exposure to continuous antigen activation can lead to antigen escape and premature exhaustion of CAR-T cells, resulting in a decline in their tumoricidal capacity.
Despite these challenges, scientists have continued to explore and innovate, achieving substantial progress in the field of CAR-T cell therapy for solid tumors. Significant advancements include the combination of CD28 and 4-1BB co-stimulatory domains, which have shown considerable potential in eliminating solid tumors. The use of immune checkpoint inhibitors, such as anti-PD-1, has improved the durability of CAR-T cells and mitigated their exhaustion. Furthermore, the engineering of CAR-T cells through diverse strategies, including immune checkpoint inhibition, combination therapy, targeting specific antigens, gene editing techniques, and physicochemical approaches, has reversed their exhaustion and significantly enhanced their antitumor efficacy. It is noteworthy that the CAR-T cell therapy targets that have been identified and subjected to clinical trials thus far—specifically, targeting PMSA for the treatment of mCRPC, MSLN for MPM, GD2 for neuroblastoma, and HER2 for ovarian and breast cancers—have demonstrated encouraging outcomes in terms of both the efficacy and therapeutic impact of CAR-T cells. These positive findings bolster the practical potential for the clinical deployment of CAR-T cell therapy in the realm of cancer treatment. Despite the existing constraints of CAR-T cell therapy in treating solid tumors, it still requires integration with radiotherapy and chemotherapy to elevate the cure rate. In cases of recurrent and refractory hematologic malignancies, CAR-T cell therapy emerges as a potential alternative for patients who have developed resistance to radiotherapy and chemotherapy. However, to this day, radiotherapy and chemotherapy continue to hold their ground as the primary treatment modalities. The journey towards the ubiquitous clinical application of CAR-T therapy still has a considerable distance to traverse.
In summary, we have ventured into new territories to address the challenges associated with CAR-T therapy in solid tumors, and some treatment methods have exhibited promising antitumor effects in clinical settings. As research deepens and technology advances, CAR-T cell therapy for solid tumors is poised to achieve greater breakthroughs in the future. Scientists will continue to explore new targets, optimize CAR structures, develop comprehensive treatment plans, and harness gene-editing technologies to enhance the efficacy and safety of CAR-T cells in solid tumor treatments. With the progression of clinical trials and the refinement of regulatory policies, CAR-T cell therapy for solid tumors is anticipated to enter the clinical application phase sooner, offering hope to a broader population of cancer patients.
We would like to express our sincere thanks to the reviewers for their professional scrutiny of this review article, as well as for their constructive comments and invaluable suggestions on the manuscript.
H.Y. and A.Y. designed the manuscript outline and provided the writing framework. Y.L. and B.Z. collaboratively conceived and wrote the manuscript, with Y.X., R.Z., L.J. and B.Y. all contributing to its composition. Y.L., B.Z. and Y.X. jointly conceived and designed the figures. Y.L. and Y.X. worked together to collect the references. R.Z., L.J. and B.Y. reviewed the content and provided revision suggestions. A.Y. and H.Y. offered professional advice and guidance for the manuscript. All authors have made contributions to the revision of the manuscript and have read and approved the submitted version.
Not applicable.
Not applicable.
This study was supported by the Natural Science Foundation of Henan Province (grant 242300421093), the 111 Project (NO. D20036) and the Scientific and Technological Innovation Team of Shaanxi Innovation Capability Support Plan (grant 2022TD-47).
The authors declare no conflict of interest.