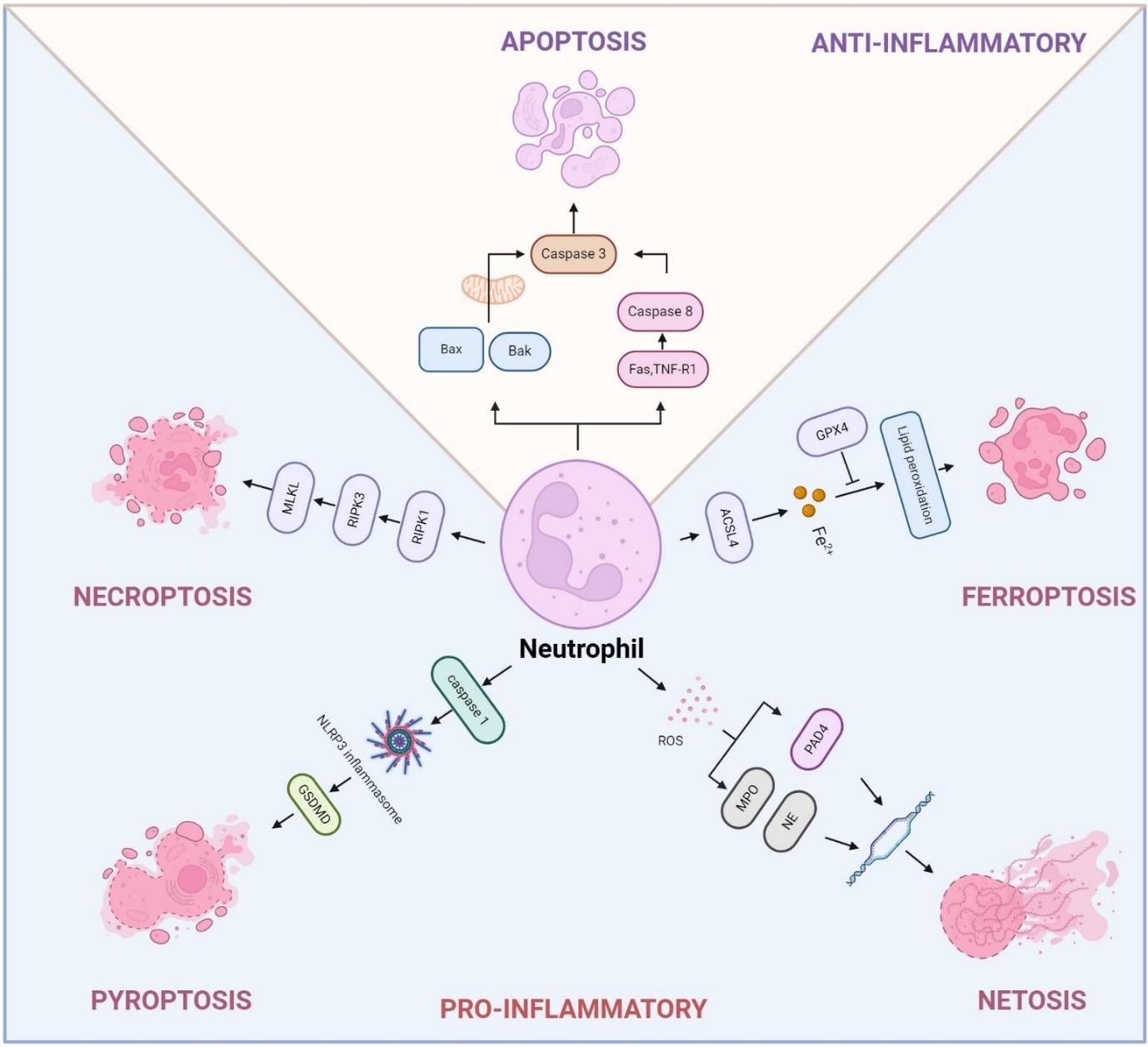
Chronic obstructive pulmonary disease (COPD) and lung cancer are closely linked, with individuals suffering from COPD at a significantly higher risk of developing lung cancer. The mechanisms driving this increased risk are multifaceted, involving genomic instability, immune dysregulation, and alterations in the lung environment. Neutrophils, the most abundant myeloid cells in human blood, have emerged as critical regulators of inflammation in both COPD and lung cancer. Despite their short lifespan, neutrophils contribute to disease progression through various forms of programmed cell death, including apoptosis, necroptosis, ferroptosis, pyroptosis, and NETosis, a form of neutrophil death with neutrophil extracellular traps (NETs) formation. These distinct death pathways affect inflammatory responses, tissue remodeling, and disease progression in COPD and lung cancer. This review provides an in-depth exploration of the mechanisms regulating neutrophil death, the interplay between various cell death pathways, and their influence on disease progression. Additionally, we highlight emerging therapeutic approaches aimed at targeting neutrophil death pathways, presenting promising new interventions to enhance treatment outcomes in COPD and lung cancer.
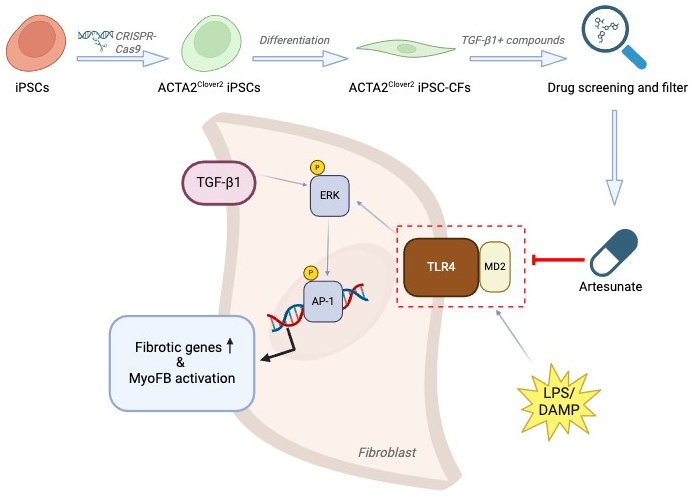
Fibrosis is a progressive pathological process that severely impairs normal organ function. Current treatments for fibrosis are extremely limited, with no curative approaches available. In a recent article published in Cell, Zhang and colleagues employed drug screening using ACTA2 reporter iPSC-derived cardiac fibroblasts and identified artesunate as a potent antifibrotic drug by targeting MD2/TLR4 signaling. This study provides new insights into strategies for exploiting existing drugs to treat fibrosis.
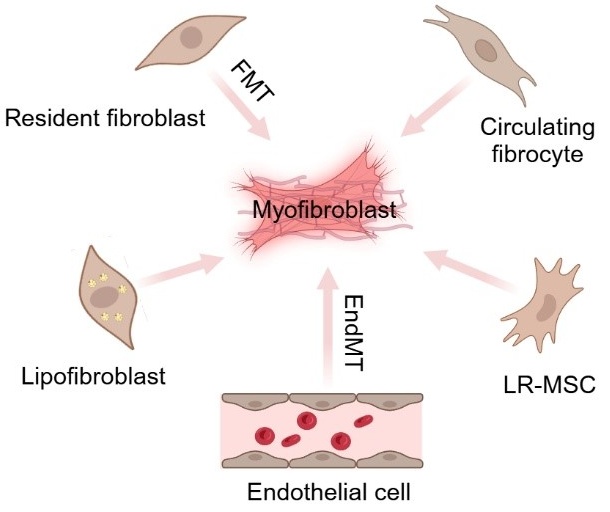
Idiopathic pulmonary fibrosis (IPF) is a progressive, irreversible, and fatal disease with an increasing incidence and limited therapeutic options. It is characterized by the formation and deposition of excess extracellular matrix proteins resulting in the gradual replacement of normal lung architecture by fibrous tissue. The cellular and molecular mechanism of IPF has not been fully understood. A hallmark in IPF is pulmonary fibroblast to myofibroblast transformation (FMT). During excessive lung repair upon exposure to harmful stimuli, lung fibroblasts transform into myofibroblasts under stimulation of cytokines, chemokines, and vesicles from various cells. These mediators interact with lung fibroblasts, initiating multiple signaling cascades, such as TGFβ1, MAPK, Wnt/β-catenin, NF-κB, AMPK, endoplasmic reticulum stress, and autophagy, contributing to lung FMT. Furthermore, single-cell transcriptomic analysis has revealed significant heterogeneity among lung myofibroblasts, which arise from various cell types and are adapted to the altered microenvironment during pathological lung repair. This review provides an overview of recent research on the origins of lung myofibroblasts and the molecular pathways driving their formation, with a focus on the interactions between lung fibroblasts and epithelial cells, endothelial cells, and macrophages in the context of lung fibrosis. Based on these molecular insights, targeting the lung FMT could offer promising avenues for the treatment of IPF.
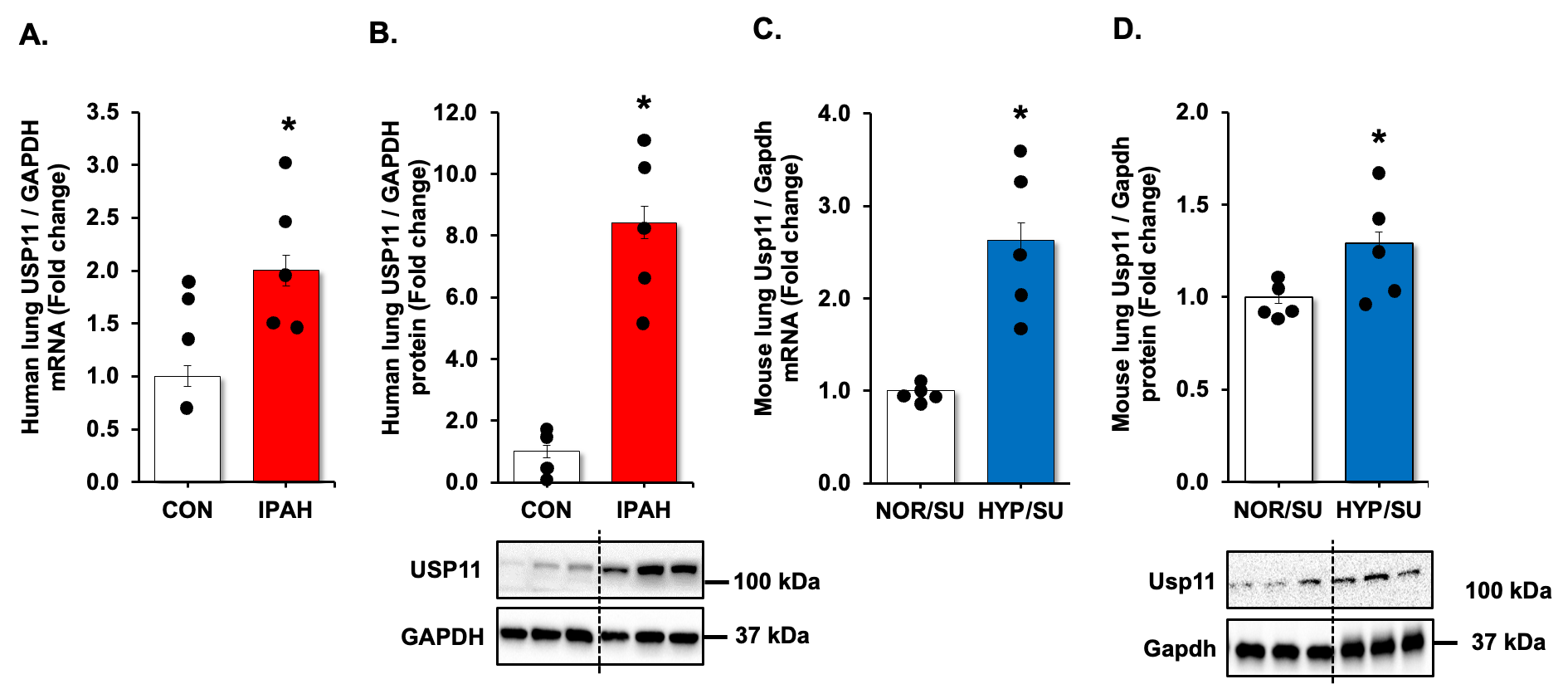
Pulmonary arterial hypertension (PAH) is a progressive, lethal, and incurable disease of the pulmonary vasculature. A previous genome-wide association study (GWAS) with Affymetrix microarray analysis data exhibited elevated histidine triad nucleotide-binding protein 3 (HINT3) in the lung samples of PAH compared to control subjects (failed donors, FD) and the positive correlations of HINT3 with deubiquitinase USP11 and B-cell lymphoma 2 (BCL2). In this study, we aim to investigate the roles and interplay of USP11 and HINT3 in the apoptosis resistance of PAH. The levels of USP11 and HINT3 were increased in the lungs of idiopathic PAH (IPAH) patients and Hypoxia/Sugen-treated mice. USP11 and HINT3 interacted physically, as shown by co-immunoprecipitation (co-IP) assay in human pulmonary arterial endothelial cells (HPAECs). HINT3 was degraded by polyubiquitination, which was reversed by USP11. Furthermore, HINT3 interacted with the anti-apoptotic mediator, BCL2. Overexpression of USP11 increased BCL2 content, congruent to elevated lung tissue levels seen in IPAH patients and Hypoxia/Sugen-treated mice. Conversely, the knockdown of HINT3 function led to a depletion of BCL2. Thus, we conclude that USP11 stabilizes HINT3 activation, which contributes to endothelial apoptosis-resistance of pulmonary arterial endothelial cells in PAH. This can potentially be a novel therapeutic target for ubiquitination modulators for PAH.