1. Introduction
Fibrosis is defined as excessive accumulation and disorganized deposition of extracellular matrix (ECM) components in tissues, following injury. Fibrosis has been shown to be a critical driver in the pathophysiology of several chronic and progressive inflammatory and metabolic diseases. It may occur in any organ or tissue, affecting single organs or multiple organs as in systemic sclerosis (SSc). The accumulation of ECM in the tissues in a disorganized fashion is the main cause of dysfunction in the affected tissue and can ultimately lead to organ failure which can be fatal. The main four vital organs that may be affected by fibrosis are lung, heart, liver and kidney—all of which have high mortality rates. Another organ which can be affected is the skin, which may not necessarily cause mortality but can lead to significant morbidity. The annual combined incidence of fibrosis-related diseases affecting these five organs is estimated to be 5000 per 100,000 people per year, causing huge disease burden. It is estimated that fibrotic diseases account for ~45% of mortality in Western countries [1]. The incidence of fibrotic diseases especially in the lung and heart are expected to increase because of COVID-19 infections [2,3].
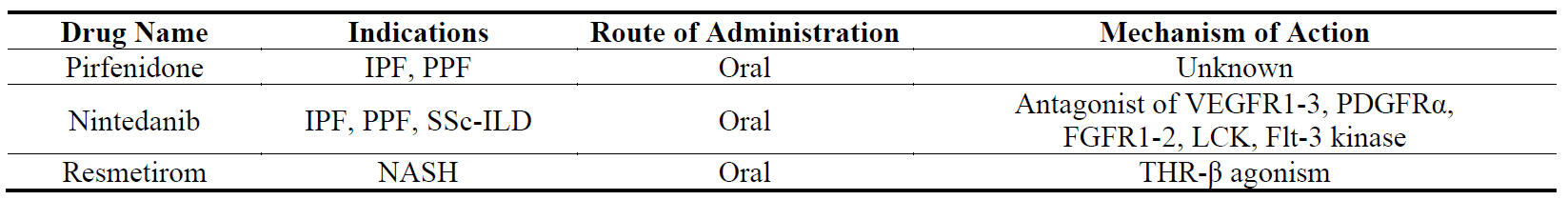
Despite these overwhelming statistics, there are only three drugs that are approved and licensed for fibrotic diseases: pirfenidone and nintedanib for lung fibrosis and resmetirom for liver fibrosis. There is certainly a lack of progress in developing novel anti-fibrotics even though the market size of fibrotic diseases is estimated to be ~$30B and several pharmaceutical companies have active R&D programmes in this field [4].
The aim of this paper is to review the current drug development activities for novel anti-fibrotic medicines in five major fibrotic conditions (lung, heart, kidney, liver and skin) and to discuss the bottlenecks and potential solutions to them.
2. Major Fibrotic Conditions
2.1. Lung Fibrosis
Fibrosis of the lung can be observed at the end stage of various parenchymal lung diseases including idiopathic pulmonary fibrosis (IPF), fibrotic interstitial lung disease (fILD) and fibrotic hypersensitivity pneumonitis (fHP) which are grouped under the umbrella term of “progressive pulmonary fibrosis” [5]. Prevalence of IPF is estimated to be 0.33–2.51 per 10,000 persons in Europe and 2.40–2.98 per 10,000 persons in North America. Prevalence of non-IPF PPF is estimated to be 0.22–2.0 per 10,000 persons in Europe and 2.8 per 10,000 persons in North America [6]. Autoimmune connective tissue disease, smoking, family history, radiation, chemotherapy, asthma, allergic airway inflammation, exposure to substances such as asbestos or silica dust, sarcoidosis and viral infections such as COVID-19 have been suggested as etiological factors for non-IPF PPF [7]. Family history has been suggested to be a contributing factor also to IPF [8]. Diagnosis of fibrosis using radiological techniques such as thin-section computerized tomography has recently been recognized as key to the appropriate categorization of pulmonary fibrosis [9].
Fibrotic lung diseases are characterized by progressive worsening of dyspnea and lung function and are associated with a poor prognosis and high mortality. The median survival in patients with IPF and non-IPF PPF without a lung transplant have been reported to range from two to five years [10,11]. The age-standardized mortality rate for IPF vary from 0.5 to 12 per 100,000 population per year with regional differences [12].
Fibrosis in the lung has been suggested to be initiated by repeated sub-clinical injury to epithelial cells and subsequent destruction of the alveolar-capillary basement membrane [13]. This process triggers an inflammatory response which results in the recruitment and infiltration of immunes cells such as macrophages, and the release of pro-inflammatory and pro-fibrotic cytokines and chemokines such as tumor necrosis factor (TNF)-α, transforming growth factor (TGF)-β, monocyte chemotactic protein (MCP)1/CCL2, macrophage inhibitory protein (MIP)1α/CCL3, and T-helper (Th)2-chemokines such as CCL17, CCL18 and CCL22 [13]. This leads to trans-differentiation (transformation) of various cell types such as fibroblasts, epithelial cells, endothelial cells, fibrocytes, mesenchymal stem cells and/or pericytes to myofibroblasts; the origin of myofibroblasts in lung fibrosis has been widely studied and debated [13]. Myofibroblasts in comparison to their cells of origin have increased proliferation and ECM production capacity and adopt the ability to contract due to their alpha smooth muscle actin (α-SMA) contractile stress fibers. In physiological wound healing, myofibroblasts are removed from the wound once the healing process is complete by undergoing apoptosis. However, in fibrosis (often referred to as aberrant wound healing), myofibroblasts lose their apoptotic potential, leading to their prolonged existence and excessive proliferation, plus the subsequent excessive accumulation of ECM proteins [14]. Under physiological conditions, the ECM not only determines the tissue architecture of the lung but also provides mechanical stability and the elastic recoil required for pulmonary function. It also regulates and controls myofibroblast transformation [15,16]. However, during fibrosis this balance is lost, and remodeling of the lung is dysregulated which leads to tissue destruction and loss of function [15].
Current clinical management of IPF is comprised of anti-fibrotic drugs (pirfenidone, nintedanib), anti-inflammatory/anti-tussive drugs (oral corticosteroids and opioids) to decrease cough and improve quality of life, anti-acids/proton pump inhibitors to reduce gastroesophageal reflux and lung transplantation [17]. Current clinical management of non-IPF PPF involves the removal of the underlying cause (e.g., limiting the occupational exposure to substances), treatment of underlying disease (e.g., immunosuppressants for systemic sclerosis, rheumatoid arthritis), anti-inflammatory/anti-tussive drugs (oral corticosteroids and opioids) and lung transplantation. The use of anti-fibrotic drugs (pirfenidone, nintedanib) in non-IPF PPF is debated [18]. Nintedanib is approved in many countries for ILD due to systemic sclerosis (SSc-ILD), but pirfenidone is not [19].
2.2. Cardiac Fibrosis
Fibrosis in the heart can be observed in several cardiovascular conditions such as heart failure (HF), hypertension, myocarditis and ischemic or non-ischemic cardiomyopathy. Fibrotic changes can be observed in the myocardium or the valves, or both [20].
Heart failure has an estimated global prevalence of 64 million and this number is projected to increase due to the ageing population. It is estimated that approximately 30–50% of patients with HF suffer from HF with preserved ejection fraction (HFpEF) which has a median survival rate of 2.1 years [21]. Although several therapies exist for HF with reduced ejection fraction (HFrEF) (e.g., beta adrenoceptor blockers, drugs targeting renin angiotensin aldosterone system, and sodium-glucose co-transporter 2 inhibitors), no treatment has been shown to be efficacious for HFpEF. The etiological and risk factors for HFpEF are age, myocardial infarction, aortic stenosis, valvular disease, diabetes, obesity, chronic kidney disease, hypertension, atrial fibrillation, chronic obstructive pulmonary disease, iron deficiency, alcohol consumption, anthracyclines and anemia [22]. The pathophysiology of HFpEF is complex and comprises several pathological mechanisms such as endothelial dysfunction, chronic inflammation, and cardiomyocyte dysfunction. Among these mechanisms, fibrosis is a common outcome regardless of etiology and all pathophysiological mechanisms converge on fibrosis [23].
The pathophysiology of cardiac fibrosis shares several features with lung fibrosis. Cardiomyocyte death after injury such as ischemia or due to other disease leads to an inflammatory response and transformation of cardiac resident fibroblasts to α-SMA-positive myofibroblasts [24]. Resident and circulating mesenchymal cell progenitors, pericytes, endothelial cells and epicardial epithelial cells have also been suggested as possible sources for myofibroblasts [24]. Pro-inflammatory and pro-fibrotic mediators released by monocytes, macrophages, lymphocytes, endothelial cells and mast cells during fibrosis include TGF-β, TNF-α, IL-6, platelet-derived growth factor (PDGF), IL-4, IL-1β, histamine, endothelin-1 (ET-1) and MCP-1. Mast cells release several proteases such as chymase and tryptase which are shown to be pro-fibrotic. Chymase has been suggested to exert fibrogenic actions through generation of angiotensin II or through activation of the TGF-β pathway. It should be noted that chymase-induced angiotensin II generation is not affected by angiotensin converting enzyme (ACE) inhibitors which may explain the progression of cardiac fibrosis despite ACE inhibition [24]. Proliferation of myofibroblasts leads to excessive accumulation of fibrillar collagen in the cardiac interstitium which is the hallmark of cardiac fibrosis. Myocardial infarction causes the death of a significant number of cardimyocytes, resulting in their replacement with disorganized fibrillar collagen containing fibrous tissue, since the adult human heart has negligible regenerative capacity [24]. Loss of functional myocardial tissue leads to ventricular stiffness and diastolic dysfunction initially, followed by cardiac remodeling, ventricular dilation and systolic failure [25]. Fibrotic ventricular remodelling can also promote arrhythmogenesis through impaired conduction and subsequent generation of re-entry circuits [26].
The current clinical management of HFpEF is comprised of management of associated conditions (i.e., diabetes, atrial fibrillation, obesity, chronic kidney disease, myocardial ischemia, hyperlipidemia), lifestyle modifications and pharmacotherapy to manage the symptoms (i.e., diuretics, sodium-glucose co-transporter 2 (SGLT2) inhibitors, mineralocorticoid receptor antagonists (MRA) and ACE inhibitors). There are no approved drugs for treatment of cardiac fibrosis.
2.3. Liver Fibrosis
Autoimmune hepatitis, biliary obstruction, iron overload, non-alcoholic fatty liver diseases (such as non-alcoholic steatohepatitis; NASH), viral hepatitis B & C and alcoholic liver disease are chronic diseases of the liver that have fibrosis in the core of their pathogenesis. Chronic liver diseases are estimated to cause 2 million deaths per year hence are the 11th leading cause of death worldwide [27]. Alcohol is the leading cause of end stage liver failure, globally and is responsible for almost 60% of cirrhosis cases in Europe, North and Latin America [28]. Another significant fibrotic liver disease is NASH which affects a quarter of the global adult population with considerable variation depending on geographic region, ethnicity, genetic variants and lifestyle factors [29]. The percentage of total deaths from all causes attributable to NASH increased from 0.10% to 0.17% in the last three decades [30].
Liver fibrosis pathophysiology is like that of lung and cardiac fibrosis but with distinct differences. The main source for myofibroblasts in the liver are hepatic stellate cells, formerly known as lipocytes, Ito cells or perisinusoidal cells [31,32]. It should be noted that myofibroblasts can also originate from cells other than hepatic stellate cells in the liver such as portal myofibroblasts and hematopoietic stem cells [33,34]. Another difference is the regenerative property of the liver. After an acute injury such as viral hepatitis, parenchymal cells can regenerate and replace the necrotic cells. However, if the injury persists such as in chronic alcohol abuse, eventually liver regeneration fails, and the hepatocytes are replaced with ECM. The third difference is the main pro-fibrotic cytokine; PDGF, which is mainly produced by Kupffer cells, is the predominant mitogen but other cytokines and chemokines such as TGF-β, TNF-α and ET-1 are also involved [35].
During the progression of fibrosis in the liver, functioning hepatocytes are replaced with stiff, disorganized ECM. Eventually collagen bands in the liver join up and lead to the formation of new vascular connections between portal fields and central veins, known as bridging fibrosis. This leads to frank cirrhosis with typical features like portal hypertension and liver failure which can progress to hepatocellular carcinoma.
The current clinical management of NASH is comprised of lifestyle modifications, anti-fibrotic drug treatment (resmetirom), other pharmacotherapy (pioglitazone, vitamin E) and bariatric surgery [36]. The current clinical management of chronic liver disease includes lifestyle modifications (i.e., dietary changes, alcohol abstinence, weight management), management of underlying disease (i.e., antivirals for viral hepatitis, immunosuppressants for autoimmune hepatitis), management of complications of cirrhosis ascites, variceal bleeding, hepatic encephalopathy and hepatorenal syndrome) and liver transplantation [37].
2.4. Kidney Fibrosis
Kidney or renal fibrosis, characterized by glomerular (called glomerulosclerosis) and interstitial (called tubulointerstitial) fibrosis, represents the final stage of chronic kidney disease (CKD). As CKD progresses, patients can advance to end-stage renal disease (ESRD), necessitating renal replacement therapies such as dialysis or transplantation. Various diseases, including glomerulonephritis, metabolic disorders like diabetes mellitus and atherosclerosis, obstructive nephropathy, interstitial nephritis, and cystic nephropathies such as polycystic kidney disease, can lead to CKD. However, kidney fibrosis is the common end result of all these conditions [38].
CKD is one of the most prominent causes of morbidity and mortality affecting an estimated 843.6 million individuals worldwide. Its prevalence is estimated to be 10.4% among men and 11.8% among women globally with significant differences due to geography and income levels [39]. The number of deaths caused by CKD doubled from 0.6 million in 1990 to 1.4 million in 2019 [40]; this increase has been attributed to an ageing population and increased incidence of diabetes mellitus.
Kidney fibrosis is similar to lung, heart and liver fibrosis and is characterized by excessive accumulation of ECM proteins by myofibroblasts. The origin of myofibroblasts in the kidney is suggested to be resident fibroblasts, pericytes, mesenchymal stem cell (MSC)-like cells, epithelial cells, endothelial cells and circulating bone marrow-derived cells [41]. The injured tubular cells and infiltrated immune inflammatory cells release profibrotic mediators such as TGF-β, PDGF, FGF2 and HER2 in the fibrotic niche [42] which leads to the transformation and activation of myofibroblasts. Other mediators such as angiotensin II (Ang-II), nuclear factor-kappa B (NFκB) and plasminogen activator inhibitor-1 (PAI-1) have also been suggested to be involved in the renal fibrotic niche. Tubular epithelium plays an important role in this process as these cells, following repetitive injury, release mediators to recruit inflammatory cells and activate myofibroblast differentiation, proliferation, and matrix secretion [42]. A unique feature of CKD is a pathology called vascular rarefaction which is a consequence of kidney fibrosis. It is caused by the apoptosis, detachment, and dysfunction of endothelial cells and characterized by a decrease in capillary density leading to ischemic and hypoxic conditions [43]. Continuous deposition of ECM results in fibrous scars and distorts the fine architecture of kidney tissues, leading to the collapse of renal parenchyma, which is comprised of tubular atrophy, capillary loss and podocyte depletion, ultimately resulting in the irreversible loss of kidney function [44].
The current clinical management of CKD includes management of co-morbidities (i.e., diabetes, hypertension and hyperlipidemia), diet, lifestyle modifications, dialysis and kidney transplantation. There are no approved anti-fibrotic drugs for CKD or kidney fibrosis [45].
2.5. Skin Fibrosis
Dermal fibrosis can be observed in a wide spectrum of skin diseases such as scleroderma, nephrogenic fibrosing dermopathy, mixed connective tissue disease, scleromyxedema, scleroderma and eosinophilic fasciitis. Exposure to chemical/physical agents or trauma to the skin are also potential causes of fibrotic skin disease such as keloid and hypertrophic scars [46,47,48].
Scleroderma can be categorized into two main types, localized (exclusive to the skin and underlying tissues) and systemic (initial skin fibrosis, followed by fibrosis of other organs). Localised scleroderma (LoS) is a rare disease, with an incidence of 0.4–2 per 100,000 individuals, with >90% of cases being diagnosed in children [49,50]. LoS is clinically characterised by the formation of ‘morphea’ on the trunk and/or limbs, which is the result of the thickening of the skin & underlying tissues, caused by excessive collagen deposition in these areas [51].
Systemic sclerosis (SSc) is a multisystem autoimmune and vascular disease of unknown etiology which results in fibrosis of various organs. The global prevalence of SSc is estimated to be 19 per 100,000 persons [52]. The commonly accepted pathogenesis of SSc involves vascular injury (i.e., capillary loss and arteriolar stenosis) and aberrant activation of vascular cells (i.e., endothelial cells) which are secondary to autoimmune attacks and unknown environmental factors. This dual vasculopathy leads to tissue hypoxia, inflammation, activation of pro-fibrotic pathways and eventual tissue fibrosis [53]. Due to multiple organ involvement, SSc has the highest mortality rate among any of the autoimmune rheumatic diseases [54]. Current clinical management of SSc is organ-based. For the skin fibrosis, a wide variety of immunosuppressants have been used, but only a few have been studied in randomized clinical trials [55].
It is estimated that 100 million people in the developed world are affected by some sort of cutaneous scarring, following burn injury, trauma, or surgery every year. The types of scars with the highest incidence are hypertrophic (incidence of 32–72%) and keloid scars (0.09–16%, depending on ethnic background). These types of scars can result in itching, pain, loss of function, contractures, disfigurement, and diminished quality of life [56,57,58]. Like other fibrotic diseases, the pro-fibrotic microenvironment secondary to tissue injury and inflammation drives the transformation of fibroblasts, fibrocytes, pericytes, smooth muscle cells and epithelial cells to myofibroblasts [59]. Current clinical management of cutaneous scars includes steroid injection, laser therapy, bleomycin or 5-fluorouracil injection, cryotherapy and surgical removal of the scar tissue [58].
3. Approved Drugs
3.1. Pirfenidone
Pirfenidone () was the first approved anti-fibrotic drug. It was first approved in Japan in 2008, followed by the EU in 2011, Canada in 2012 and the USA in 2014 and is now available as a generic medication for the treatment of IPF. It was first reported in patents in the 1970s. The demonstration of its anti-fibrotic properties in a hamster model of bleomycin-induced lung fibrosis in the 1990s provided support for its development to treat IPF [60]. Its exact molecular target is unknown but several targets such as TGFβ, TNF, IL-10, p38α/γ and MRC5 have been suggested [60].
Table 1. The names, indications, route of administration and mechanisms of action of the drugs that have been approved for fibrotic diseases.
Pirfenidone has been shown to inhibit the production and release of pro-fibrotic and pro-inflammatory cytokines such as TGF-β, TNF-α and IL-6 and to prevent TGF-β-induced transformation of lung fibroblasts to myofibroblast [60]. In both bleomycin-induced and transplant-induced lung fibrosis models pirfenidone has been shown to reduce fibrosis and dysfunction [61].
Pirfenidone has been studied in several clinical trials with IPF patients. A recent systematic review and meta-analysis in 2021 of nine randomized controlled trials showed that pirfenidone has been beneficial to prolong the progression-free survival and improve lung function in patients with IPF. Main side effects of the drug are gastrointestinal reactions, photosensitivity and skin rashes [62,63]. Pirfenidone has been recommended to be administered in adults in titration starting with 801 mg/day reaching up to 2403 mg/day in two weeks [64].
Pirfenidone has also been tested in vitro and in vivo for other fibrotic conditions which were mostly positive [60]. However, these pre-clinical results have not been translated to clinics.
3.2. Nintedanib
Nintedanib () was the second approved anti-fibrotic drug. It was approved for the treatment of IPF in 2014 and for fILD in 2020. It was discovered during a compound screening campaign to identify inhibitors of VEGF receptor 2 (VEGFR2) with the aim of developing angiogenesis inhibitors for the treatment of cancer [65]. Nintedanib targets pro-angiogenic and pro-fibrotic pathways mediated by the VEGFR family, the fibroblast growth factor receptor (FGFR) family, the platelet-derived growth factor receptor (PDGFR) family, as well as lymphocyte-specific protein tyrosine kinase (Lck) and Flt-3 kinases [65].
Nintedanib has been shown to inhibit PDGF- and VEGF-induced proliferation and motility of human lung fibroblasts. It also prevents TGF-β-induced transformation of lung fibroblasts to myofibroblasts, leading to a reduction in TGF-β-stimulated collagen secretion and deposition. Nintedanib was found to have no effect on epithelial-mesencyhmal transition (EMT) of human alveolar type II epithelial cells, and did not induce apoptosis in human lung fibroblasts [66]. In bleomycin- and silica-induced lung fibrosis models in vivo, nintedanib reduced both fibrosis and inflammation in a preventive model and to a lesser extent in therapeutic model [66].
Like pirfenidone, nintedanib has been investigated in several randomized clinical trials. A meta-analysis of these trials in 2022 demonstrated that nintedanib reduced the rate of decline by ~50% in lung function across subjects with different forms of lung fibrosis [67]. A more recent meta-analysis showed a reduction in risk of death compared to placebo in IPF and other forms of progressive pulmonary fibrosis [68]. A meta-analysis of the safety of the drug has shown that it was associated with a higher risk of adverse events especially diarrhea, nausea, vomiting and weight loss, but it was also associated with a lower risk of cough and dyspnea in IPF and fibrotic-ILD patients [69]. Nintedanib has been recommended to be administered in adults at 300 mg daily [64].
Nintedanib has been shown to reduce fibrosis in animal models of other organs such as liver and kidney in both preventive and therapeutic models [70,71], but these results have not been translated to clinic.
3.3. Resmetirom
Resmetirom has recently been approved for the treatment of non-cirrhotic NASH (). It is a selective thyroid receptor beta (THR-β) agonist. It was discovered through a compound screening campaign to identify selective THR-β agonists. THR-β agonists are known to reduce hepatic lipid levels in metabolic disorders.
Another THR-β agonist, CS271011, has been shown to lower the levels of serum triacylglycerol and cholesterol without causing liver injury, to alleviate liver steatosis and hepatic lipid accumulation, and to improve hepatic biochemical indices in a diet-induced obese (DIO) mouse model [72]. It should be noted that this particular model does not produce liver fibrosis. In DIO-NASH animal models, where the animals develop liver fibrosis, resmetirom has been shown to reduce liver weight, hepatic steatosis, plasma alanine aminotransferase activity, liver and plasma cholesterol, blood glucose & liver α-SMA expression, and to down-regulate the genes involved in fibrogenesis [73].
In a Phase 2 trial of adults with NASH, resmetirom has been shown to reduce hepatic fat and increased the resolution of liver fibrosis [74]. In two Phase 3 trials, resmetirom was shown to reduce hepatic fat, liver stiffness/fibrosis and biomarkers of liver fibrosis and injury [75,76]. Resmetirom has been recommended to be administered in adults at 80–100 mg daily [75,76].
4. Anti-Fibrotic Compounds under Development
The compounds which are currently in clinical development are discussed in this section. Any trials with solo treatment with pirfenidone, nintedanib or resmetirom are not included. The trials are grouped according to their target class and then ranked according to their progress status (from the most to the least advanced). Where a positive result from a Phase 2/3/4 study is reported, the peer-reviewed publication that supports that conclusion is quoted where possible. Any negative result or terminated trial, unless it is published, is noted as it is stated at Clinical Trials Gov website.
4.1. Compounds Targeting Cytokines and Their Pathways
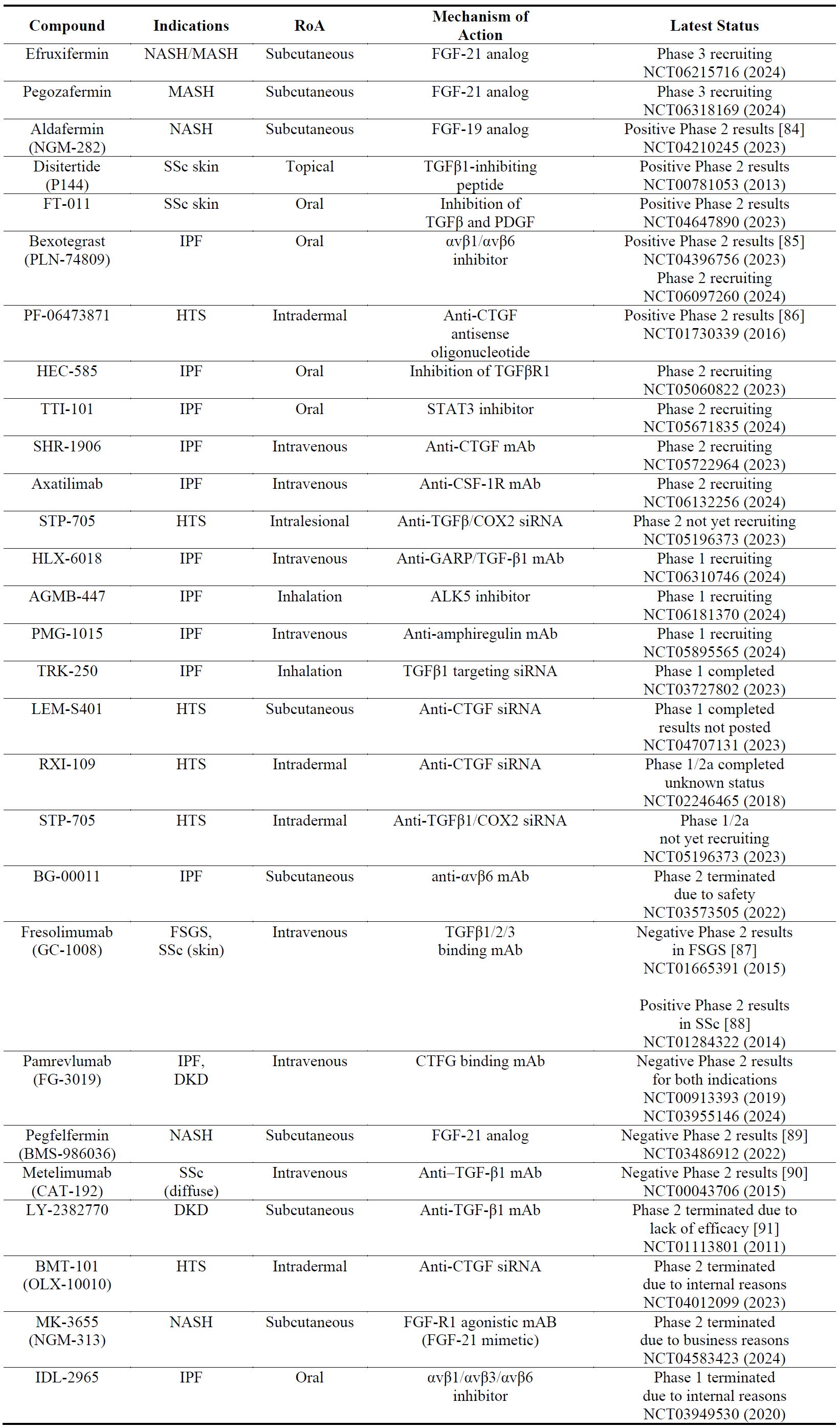
As several pro-fibrotic growth factors are involved in the pathophysiology of fibrosis, it is not surprising to observe that several compounds that are being developed, are antagonists of pro-fibrotic factors (e.g., TGF) or agonists of anti-fibrotic growth factors (e.g., FGF) (). Among the growth factors, the most frequently trialed target is TGF-β which carries certain challenges in therapeutic translation as discussed by others [77]. Foremost is the risk of systemic inhibition of TGF-β response which is required for physiological homeostasis (e.g., wound healing, immune response, vascular effects). Therefore, upstream tissue specific targets such as integrins [78] and downstream fibrotic-tissue specific targets like CTGF [79] are pursued. Among the FGF subtypes, FGF-19 [80] and FGF-21 [81] are the choice of targets in fibrosis. The list of trials in the growth factors space () reveals that the FGF-21 analogs efruxifermin and pegozafermin are the most advanced drug candidates; this could be attributed to the suggestion that FGF-21 ameliorates fibrosis by multiple mechanisms [82]. It should be noted that most of the FGF-21 analogs have been developed for the treatment of obesity and T2DM [83] hence their use in NASH and MASH.
Table 2. The names, indications, route of administration (RoA), mechanisms of action and the latest status of the compounds that target growth factors. The year next to Clinical Trial number (NCT) denotes the date of the last available update. DKD, diabetic kidney disease; FSGS, focal segmental glomerulosclerosis; HTS, hypertrophic scarring; IPF, idiopathic pulmonary fibrosis; mAb, monoclonal antibody; MASH, metabolic dysfunction-associated steatohepatitis; NASH, non-cirrhotic nonalcoholic steatohepatitis; SSc, systemic sclerosis.
4.2. Compounds Targeting Kinases
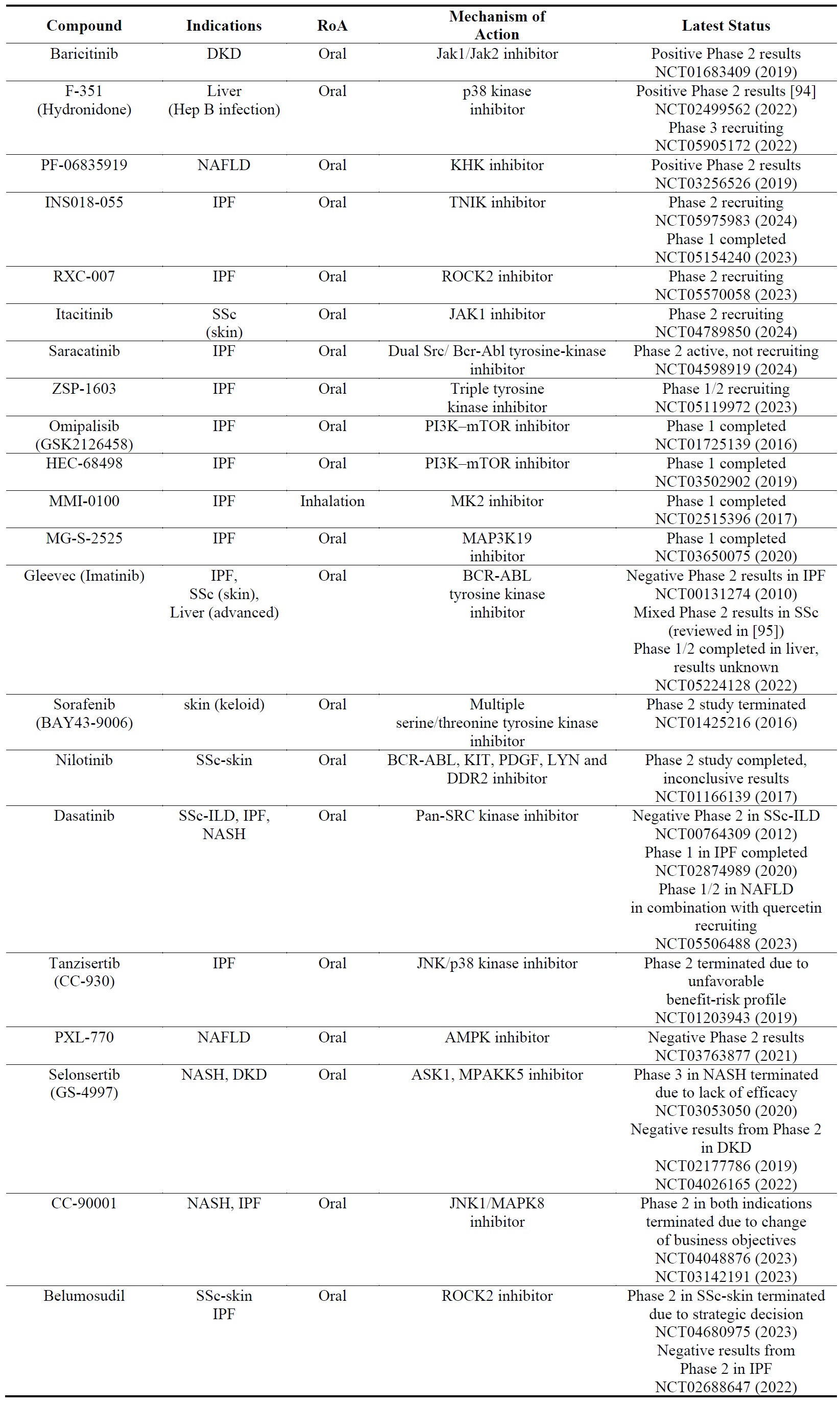
A multitude of pro-fibrotic mediators such as TGFβ, CTGF, PDGF and FGF involve several kinases in their signalling cascades and their inhibition has been shown to be anti-fibrotic in preclinical models [92]. Kinases in general are druggable targets and most of the kinase inhibitors are orally bioavialable making them ideal drug candidates. Some of these kinase inhibitors work dually by inhibiting fibrotic pathways and also inflammatory pathways which can be a pro-fibrotic driver. Most of the kinase inhibitors in were originally developed for cancer or auto-immune disease and out of 500 kinases encoded by the human genome, only 50 have so far been targeted for those therapeutic areas [93]. It is therefore predicted that more kinase inhibitors will be developed in the future. However, the overall safety profile of kinase inhibitors, particularly for non-cancer indications, still needs to be improved. The results from Phase 2 studies in fibrosis () is mixed, some kinase inhibitors (i.e., baricitinib) seem to be progressing while others (i.e., selonsertib and tanzisertib) have efficacy and safety issues. No kinase inhibitor has progressed to Phase 3 yet.
Table 3. The names, indications, route of administration (RoA), mechanisms of action and the latest status of the compounds that target kinases. The year next to Clinical Trial number (NCT) denotes the date of the last available update. DKD, diabetic kidney disease; JAK, janus kinase; KHK, ketohexokinase; TNIK, TRAF2 and NCK-interacting protein kinase; ROCK2, Rho associated coiled-coil containing protein kinase 2; PI3K, phosphoinositide 3-kinase; mTOR, mammalian target of rapamycin; MK2, MAP kinase-activated protein kinase 2; MAP3K19, mitogen-activated protein kinase 19; AMPK, 5′ adenosine monophosphate-activated protein kinase; DDR2, discoidin domain receptor 2; JNK, c-Jun N-terminal kinase; ASK1, apoptosis signal-regulating kinase 1; MPAKK5, MAPK activated protein kinase 5; IPF, idiopathic pulmonary fibrosis; mAb, monoclonal antibody; NAFLD, non-alcoholic fatty liver disease; NASH, non-cirrhotic nonalcoholic steatohepatitis; SSc, systemic sclerosis.
4.3. Compounds Targeting PPARs
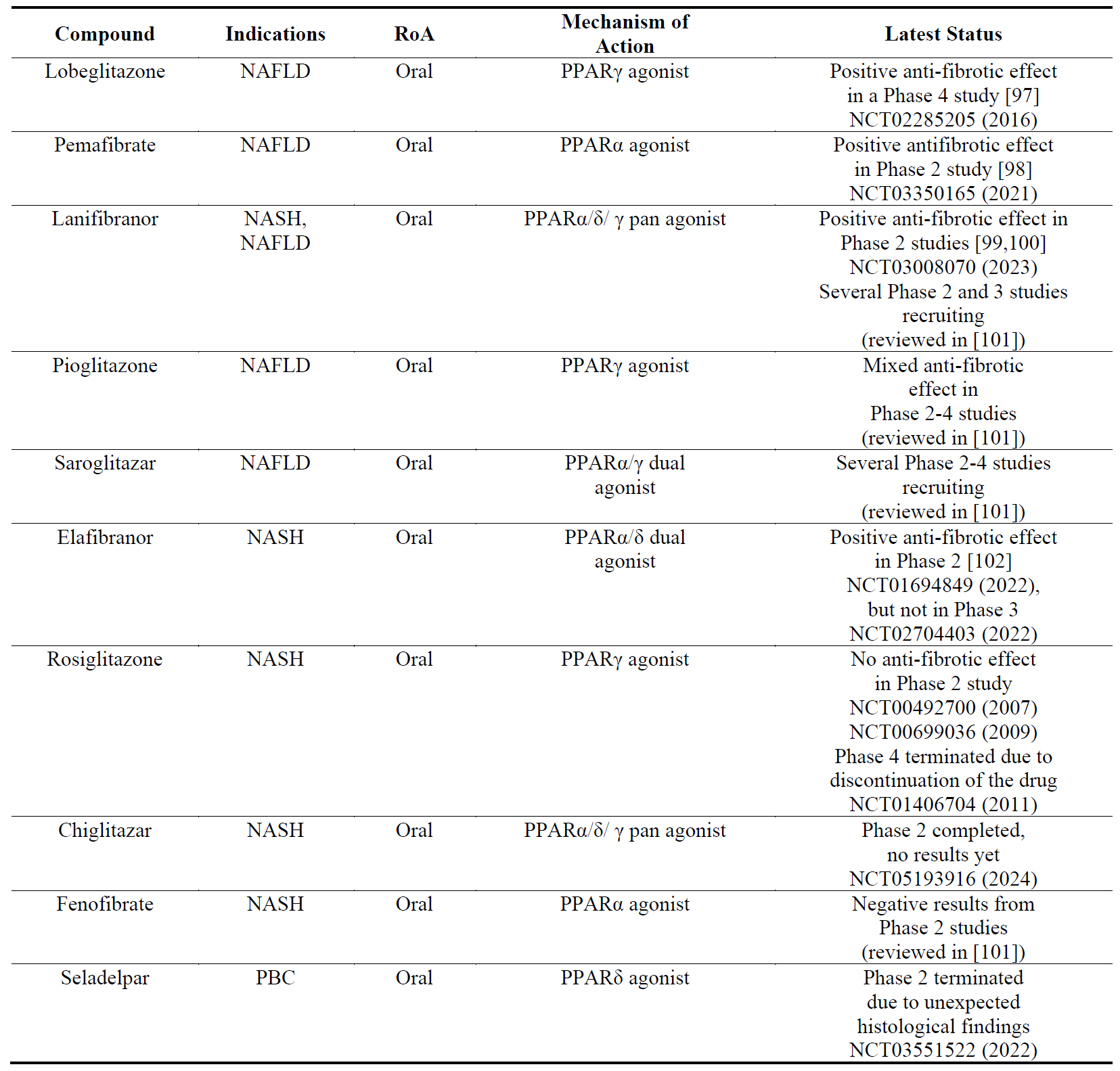
NASH/NAFLD are multi-system metabolic diseases affecting cardiovascular and renal systems and increasing the incidence of T2DM and hepatic cancer. Agonists of peroxisome proliferator-activated receptors (PPARs) have the potential to treat all these aspects of NASH/NAFLD. PPARs have three subtypes (PPARα, PPARδ(/β), PPARγ) whose agonists have been recognized to lower lipid levels and increase insulin sensitivity. PPARγ agonists have been approved for the treatment of T2DM. PPAR agonists have been shown to reduce or prevent liver fibrosis in pre-clinical models [96]. Among the drug candidates listed in , lobeglitazone is the most advanced while its long-term safety remains to be elucidated.
Table 4. The names, indications, route of administration (RoA), mechanisms of action and the latest status of the compounds that target peroxisome proliferator-activated receptors (PPARs). The year next to Clinical Trial number (NCT) denotes the date of the last available update. NAFLD, non-alcoholic fatty liver disease; NASH, non-cirrhotic nonalcoholic steatohepatitis; PBC, primary biliary cholangitis.
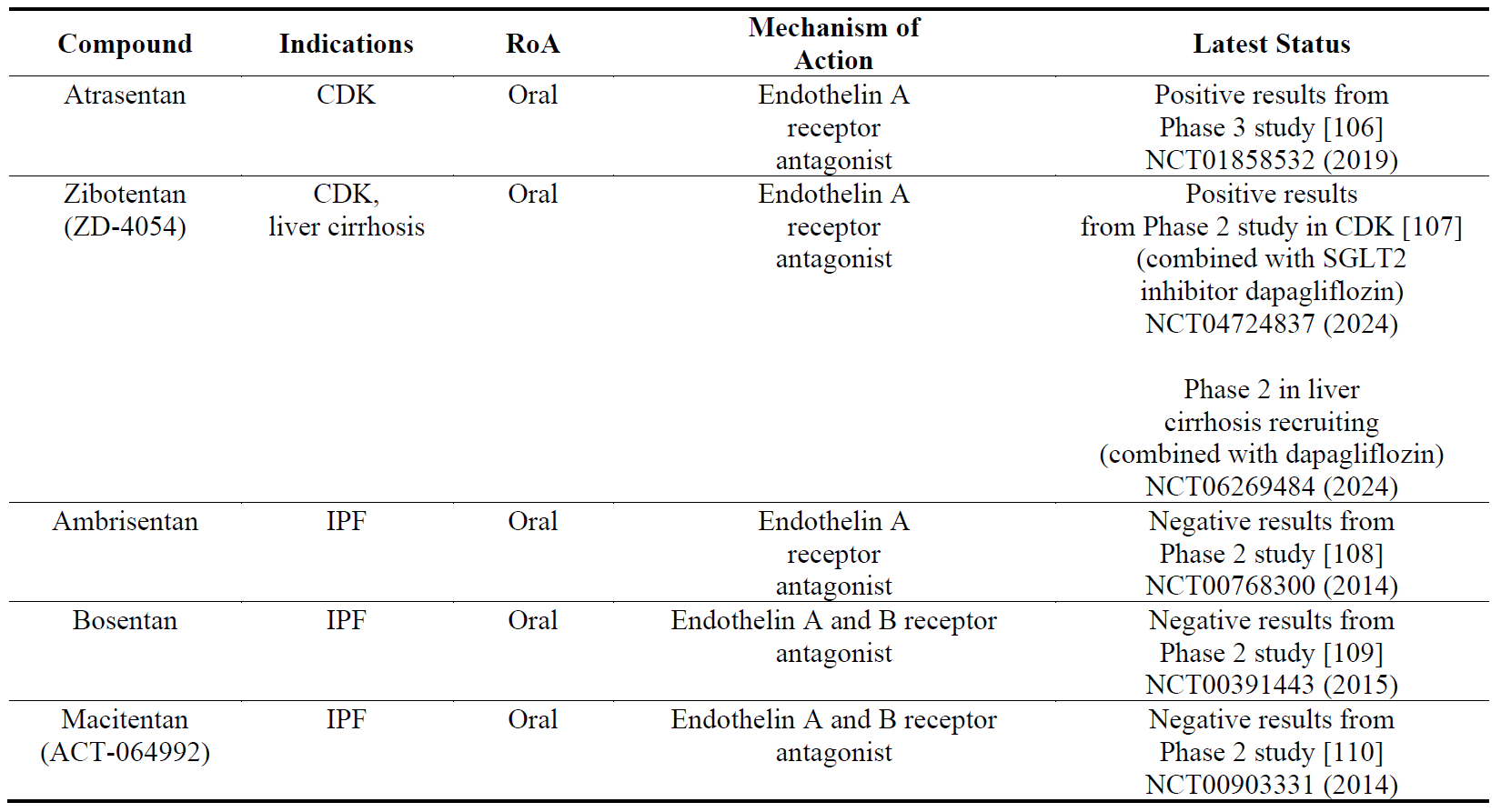
4.4. Compounds Targeting Endothelin Receptors
Endothelin receptors (type A and B) are expressed on vascular and pulmonary smooth muscle cells in the cardiovascular system, lungs and kidneys [103]. Endothelin receptor antagonists have been shown to be vasodilators and anti-fibrotic. Bosentan, a dual ET-A and -B antagonist has been approved for the treatment of pulmonary artery hypertension. In preclinical models, bosentan has been shown to be anti-fibrotic in the lungs [104]. In clinical trials () however endothelin receptor antagonists have failed show clinical efficacy in IPF which has been further confirmed by meta-analysis [105]. Attention has now seemed to turn to kidney and liver fibrosis with some positive results.
Table 5. The names, indications, route of administration (RoA), mechanisms of action and the latest status of the compounds that target endothelin receptors. The year next to Clinical Trial number (NCT) denotes the date of the last available update. CDK, chronic kidney disease.
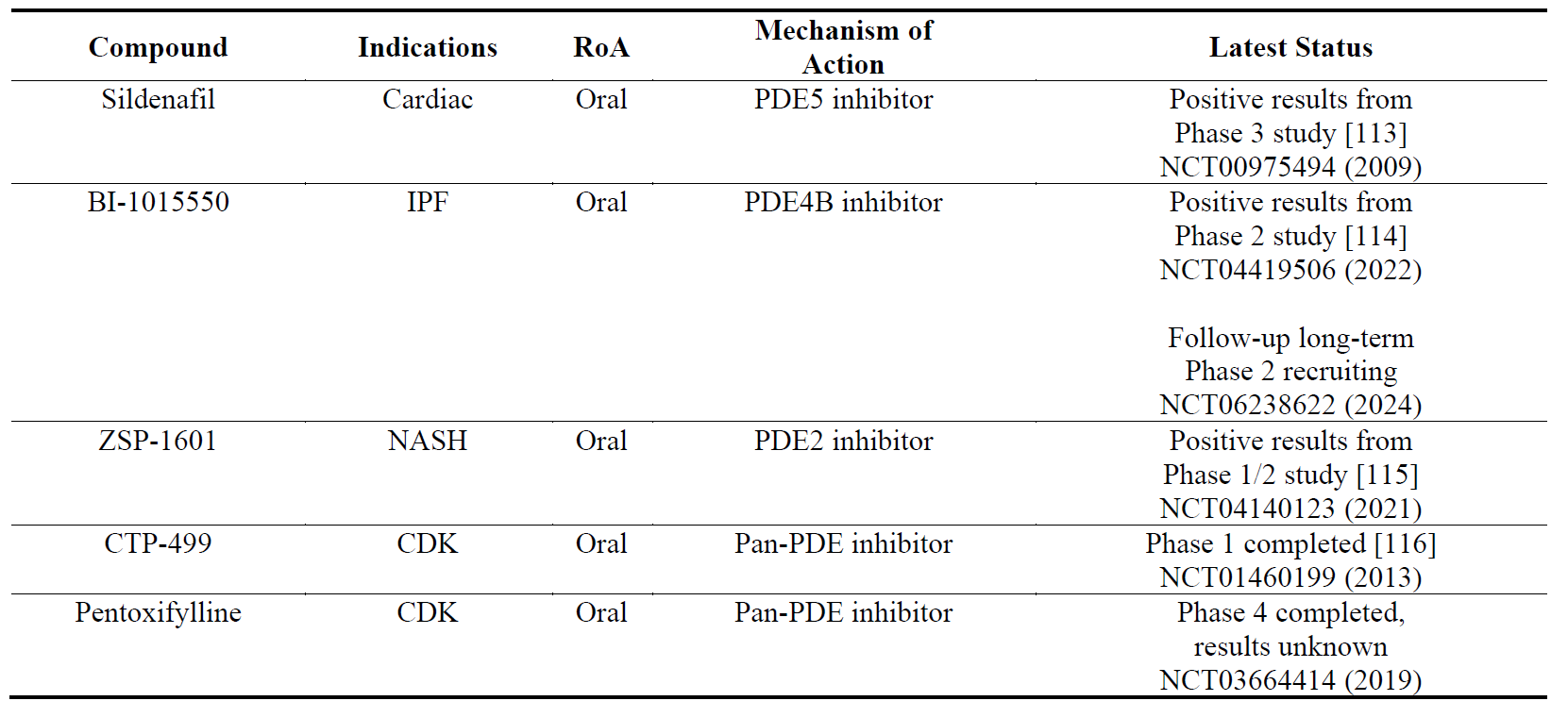
4.5. Compounds Targeting Phosphodiesterases
Phosphodiesterases (PDEs) regulate the intracellular concentrations of second messenger cyclic nucleotides cyclic adenosine monophosphate (cAMP) and/or cyclic guanosine monophosphate (cGMP). There are 11 types of PDEs with differential affinity for cAMP and/or cGMP. There have been previous reports stating that increasing cAMP or cGMP levels may have anti-fibrotic effect in preclinical models [111,112]. PDE inhibitors, which elevate intracellular cyclic nucleotide levels, therefore have been pursued as anti-fibrotic agents. There have been some positive results with the PDE5 inhibitor sildenafil (which elevates cGMP levels and is approved for the treatment of erectile dysfunction, pulmonary artery hypertension and lower urinary tract symptoms) in cardiac fibrosis and with PDE2 and PDE4B inhibitors (which elevate cAMP levels) in NASH and IPF, respectively ().
Table 6. The names, indications, route of administration (RoA), mechanisms of action and the latest status of the compounds that target phosphodiesterases (PDEs). The year next to Clinical Trial number (NCT) denotes the date of the last available update. IPF, idiopathic pulmonary fibrosis; NASH, non-alcoholic steatohepatitis; CDK, chronic kidney disease.
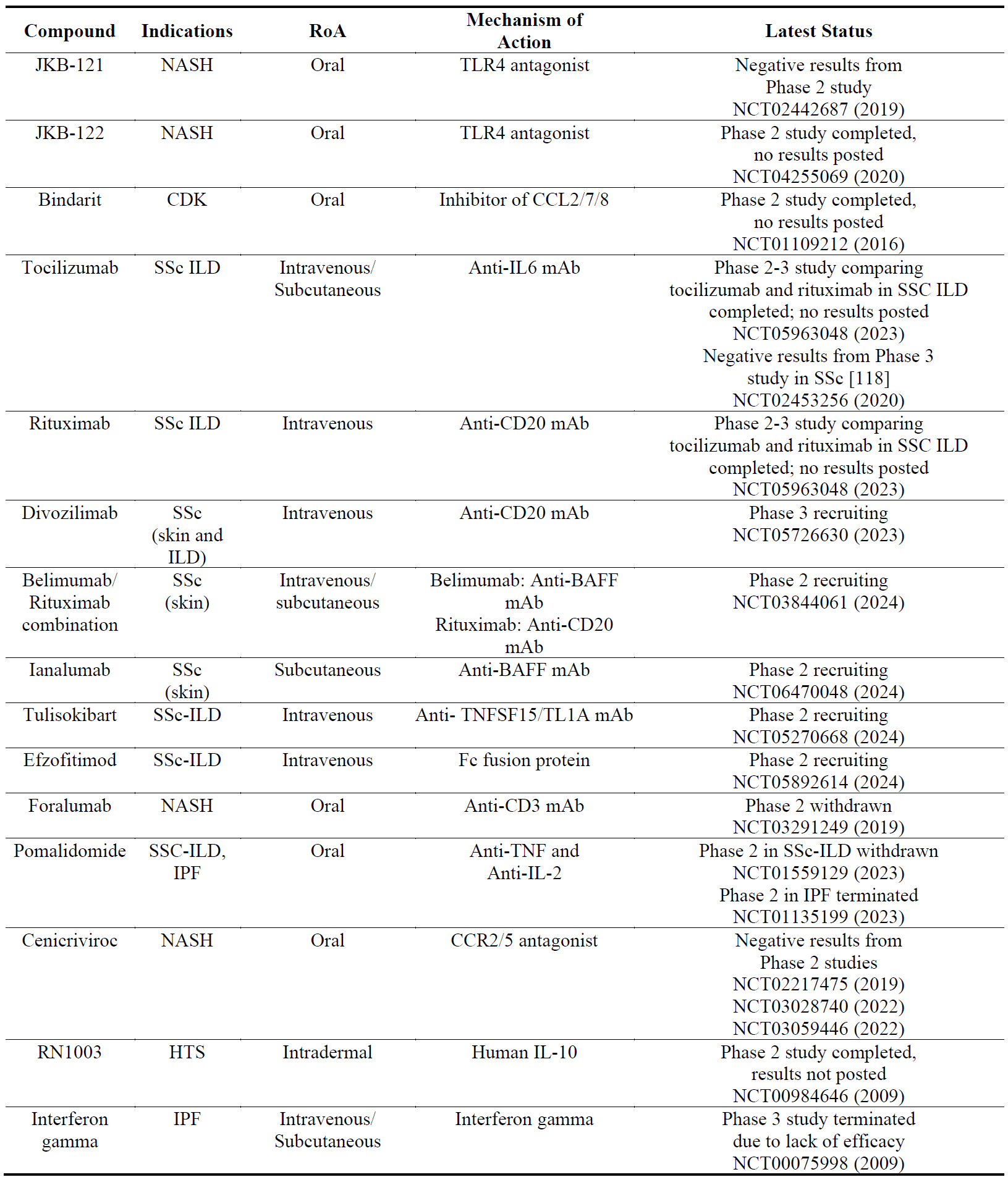
4.6. Compounds Targeting the Immune System
Both innate and adaptive immune systems play important roles in the pathophysiology of fibrosis [117]. Several drug candidates which were initially developed for auto-immune/inflammatory diseases have been trialed for fibrotic diseases (). While most of these studies have reported negative results, the most advanced drug candidate is currently an anti-CD20 mAb (divozilimab) for SSc.
Table 7. The names, indications, route of administration (RoA), mechanisms of action and the latest status of the compounds that target immune system. The year next to Clinical Trial number (NCT) denotes the date of the last available update. NASH, non-alcoholic steatohepatitis; CDK, chronic kidney disease; TLR4, toll-like receptor 4; CCL, chemokine (C-C motif) ligand; IL, interleukin; SSc, systemic sclerosis; ILD, interstitial lung disease; mAb, monoclonal antibody; BAFF, B-cell activating factor; CD, cluster of differentiation; TNF, tumor necrosis factor; TNFSF, TNF superfamily; CCR, C-C chemokine receptor.
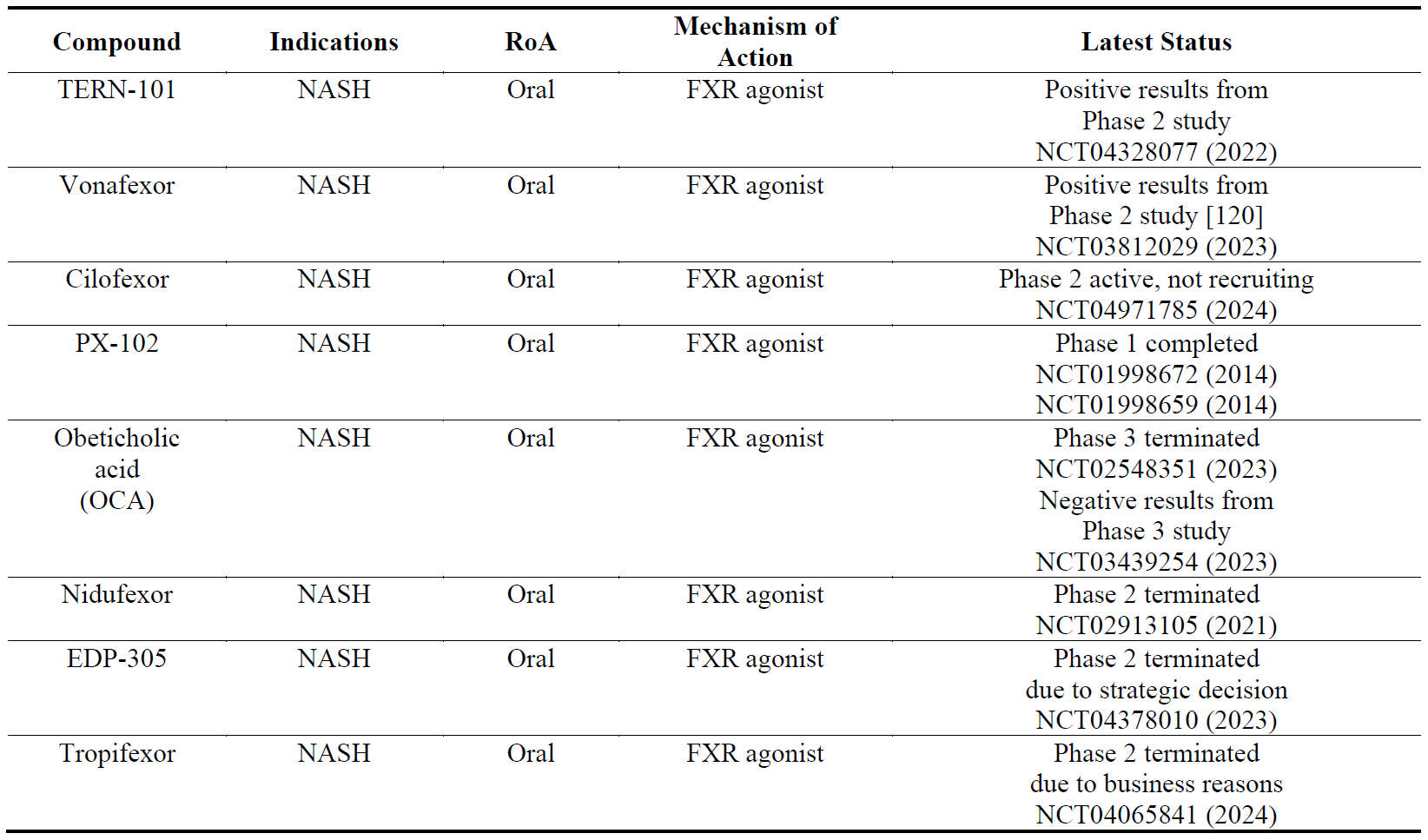
4.7. Compounds Targeting FXR
The farnesoid X receptor (FXR) is involved in regulation of several physiological mechanisms in the human gut such as bile acid synthesis and enterohepatic circulation, lipid and glucose metabolism, inflammation, fibrosis, gut barrier integrity and intestinal microbiota [119]. Since these mechanisms are involved in the pathogenesis of NASH, several FXR agonists have been developed (). Among them TERN-101 and vonafexor have been reported to be the most advanced.
Table 8. The names, indications, route of administration (RoA), mechanisms of action and the latest status of the compounds that target farnesoid X receptor (FXR). The year next to Clinical Trial number (NCT) denotes the date of the last available update. NASH, non-alcoholic steatohepatitis.
4.8. Compounds Targeting Prostaglandins
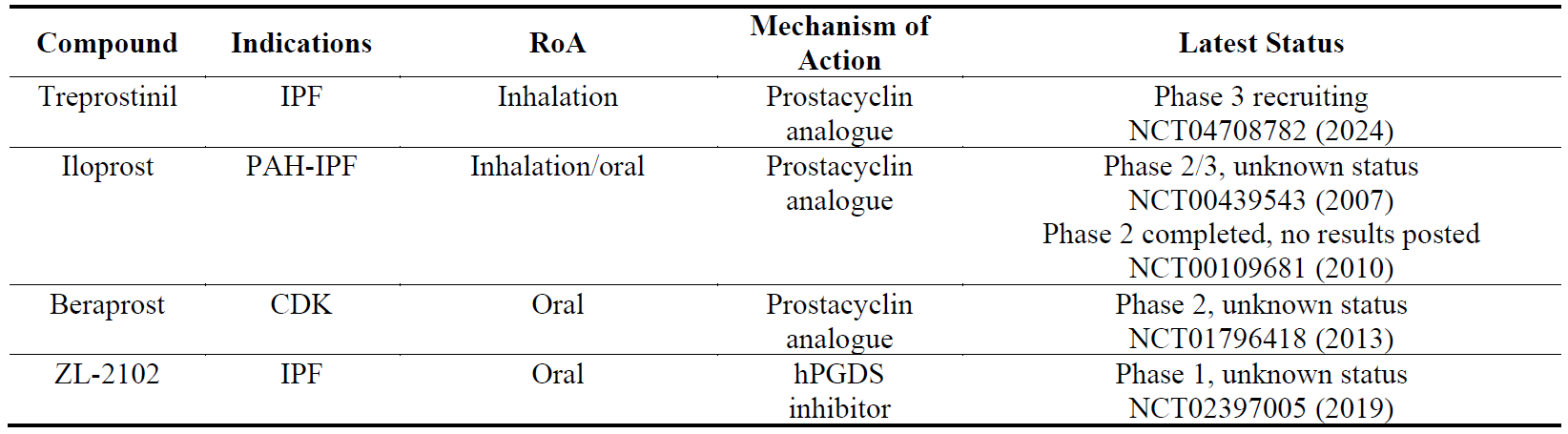
Prostaglandins are synthesized from the lipid arachidonic acid, which is present in all cellular membranes, by cyclooxygenase 1 and 2 (COX-1 and COX-2) and prostaglandin synthases [121]. Among the four prostaglandins, prostaglandin I2 (PGI2, prostacyclin) has been shown to exert anti-fibrotic effects in preclinical models of lung and kidney fibrosis [122,123], with subsequent clinical trials taking place in IPF and CDK with prostacyclin analogs (). Treprostinil, one of the inhalation formulations of prostacyclin, is the most advanced among this group. In contrast to prostacyclin, prostaglandin D2 (PGD2) is a pro-inflammatory and pro-fibrotic mediator which is synthesized by hematopoietic PGD synthase (hPGDS) [124]. ZL-2102, a selective hPGDS inhibitor has been developed for COPD, asthma and IPF [125] ().
Table 9. The names, indications, route of administration (RoA), mechanisms of action and the latest status of the compounds that target prostaglandins. The year next to Clinical Trial number (NCT) denotes the date of the last available update. IPF, idiopathic pulmonary fibrosis; PAH, pulmonary arterial hypertension; CDK, chronic kidney disease; hPGDS, hematopoietic prostaglandin D synthase.
4.9. Anti-Diabetic Compounds
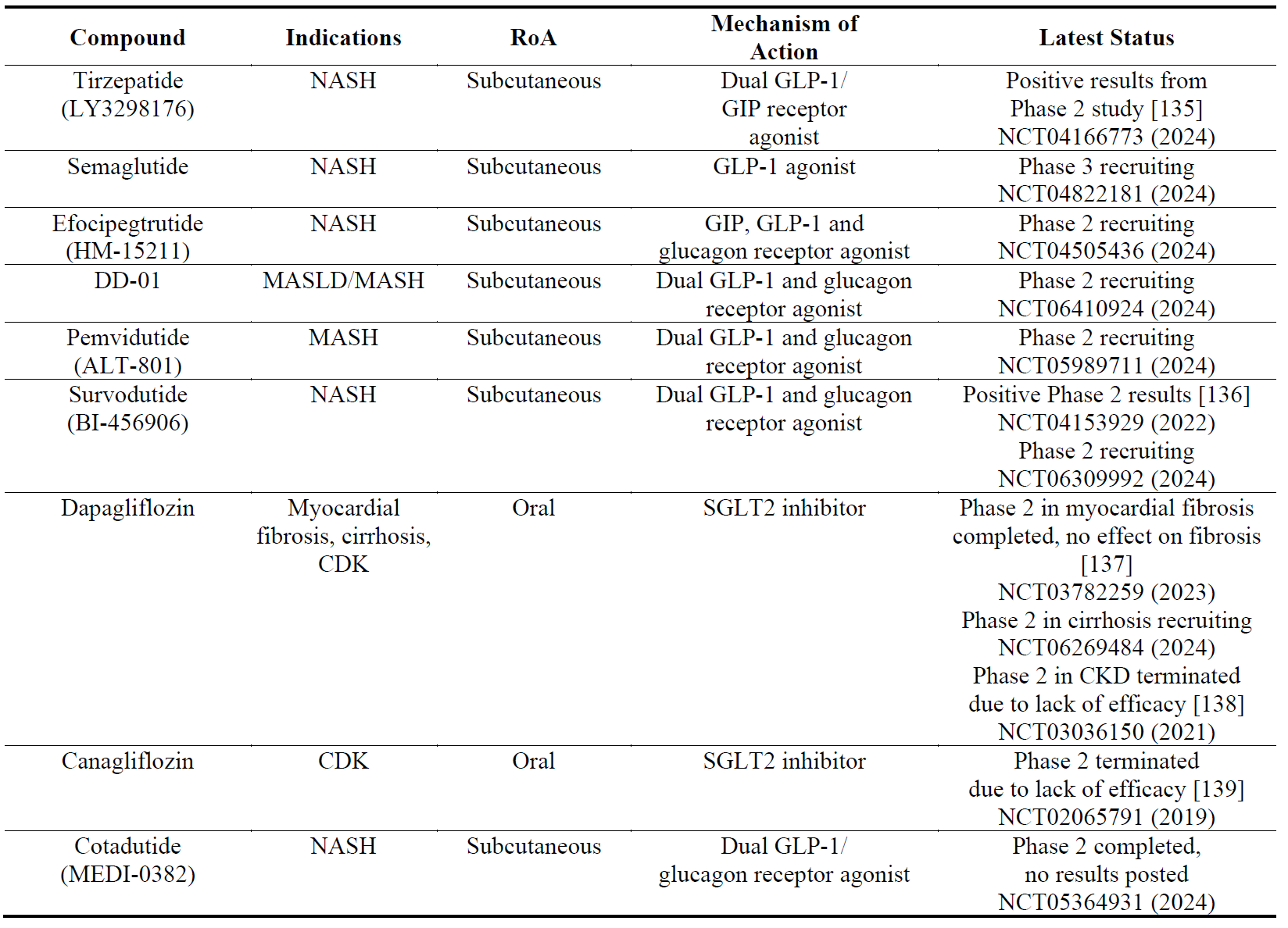
There are currently several therapeutics marketed for type 2 diabetes mellitus (T2DM): dipeptidyl peptidase-4 inhibitors (DPP-4is, gliptins), glucagon-like peptide-1 receptor agonists (GLP-1RAs) and sodium-glucose cotransporter type 2 inhibitors (SGLT2s, gliflozins). Glucose-dependent insulinotropic polypeptide (GIP) receptor agonists have also been developed but have not reached the market. Recently compounds that have dual activity have been developed [126] such as tirzepatide (LY3298176) [127], survodutide (BI-456906) [128] and pemvidutide (ALT-801) [129] which have agonistic activity on the GIP receptor and the GLP-1 receptor; and DD-01 [130] and cotadutide [131] which have agonistic activity on the GLP-1 receptor and the glucagon receptor. There have also been development of triple agonists (GIP/GLP-1/glucagon receptor) such as efocipegtrutide (HM-15211) [132], LY3437943 [133] and SAR-441255 [134]. Most of these compounds have shown efficacy in improving fibrosis markers in clinical trials of liver fibrosis, but not in kidney or cardiac fibrosis ().
Table 10. The names, indications, route of administration (RoA), mechanisms of action and the latest status of anti-diabetic compounds. The year next to Clinical Trial number (NCT) denotes the date of the last available update. NASH, non-alcoholic steatohepatitis; MASH; metabolic dysfunction-associated steatohepatitis; MAFLD, metabolic dysfunction–associated fatty liver disease; GLP, glucagon-like peptide; GIP, glucose-dependent insulinotropic polypeptide; SGLT2, sodium-glucose transport protein 2; CDK, chronic kidney disease.
4.10. Other Compounds
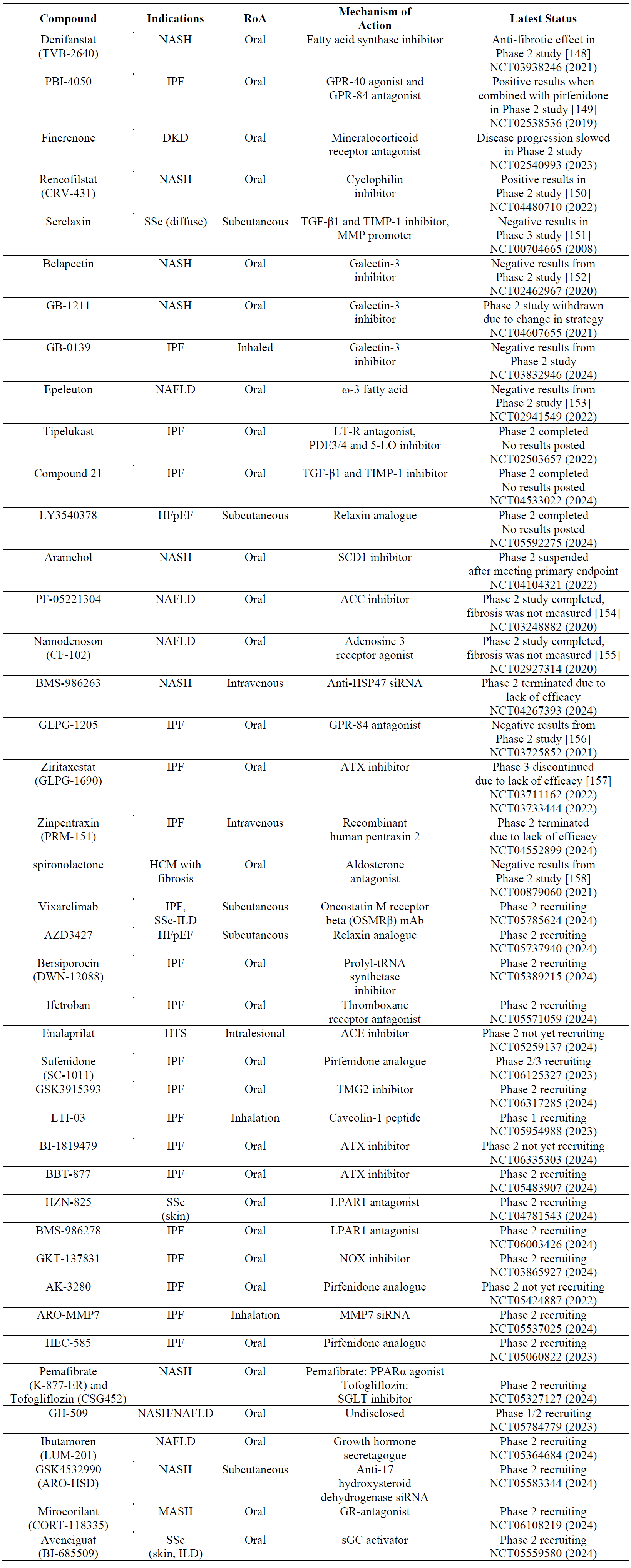
There have been several clinical trials with other compounds, most of which have given negative results (). Positive results were seen in Phase 2 studies with a fatty acid synthase inhibitor (denifanstat) in NASH, a dual GPR-40/80 antagonist (PBI-4050) in IPF, a mineralocorticoid receptor antagonist (finerenone) in DKD and a cyclophilin inhibitor (rencofilstat) in NASH (). There are also several Phase 1-2-3 studies which are recruiting with various targets ranging from NADPH-oxidase to autotaxin ().
ECM homeostasis is maintained by a fine balance between production and metabolism of ECM components which is controlled by matrix metalloproteinases (MMPs) and their inhibitors, tissue inhibitors of metalloproteinases (TIMPs). Regulation of MMPs and TIMPs have been attractive strategies for the development of anti-fibrotic drugs. MMPs and TIMPs are often co-expressed in response to multiple stimuli including inflammatory cytokines, growth factors, glucocorticoids or retinoids [140,141]. Some of the candidate molecules listed in this section either directly act on MMP/TIMPs (e.g., MMP7 siRNA) or indirectly by regulating their expression such as recombinant human gene-2 relaxin (serelaxin) [142], angiotensin type 2 receptor agonists [143,144], lysophosphatidic acid receptor 1 antagonists [145], autotaxin antagonists [146] and angiotensin cobverting enzyme inhibitors [147].
Table 11. The names, indications, route of administration (RoA), mechanisms of action and the latest status of other compounds. The year next to Clinical Trial number (NCT) denotes the date of the last available update. NASH, non-alcoholic steatohepatitis; IPF, idiopathic pulmonary fibrosis; GPR, G-protein coupled receptor; DKD, diabetic kidney disease; NAFLD, non-alcoholic fatty liver disease; LT, leukotriene; PDE, phosphodiesterase; 5-LO, 5-lipoxygenase; SCD1, stearoyl CoA desaturase 1; ACC, acetyl-CoA carboxylase; HSP, heat shock protein; ATX, autotaxin; SSc, systemic sclerosis; ILD, interstitial lung disease; HTS, hypertrophic scarring; ACE, angiotensin converting enzyme; tRNA, transfer RNA; HCM, hypertrophic cardiomyopathy; TMG2, tissue transglutaminase 2; LPAR, lysophosphatidic acid receptor; NOX, NADPH oxidase; MMP, matrix metalloproteinase; PPAR, peroxisome proliferator-activated receptor; SGLT, sodium glucose transport protein; GR, glucocorticoid receptor; sGC, soluble guanylate cyclase.
5. Analysis of Clinical Studies
5.1. Distribution According to the Organ
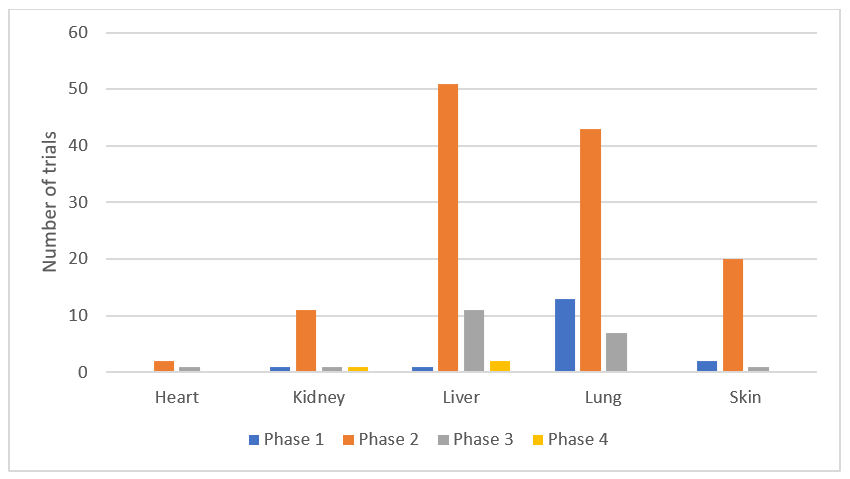
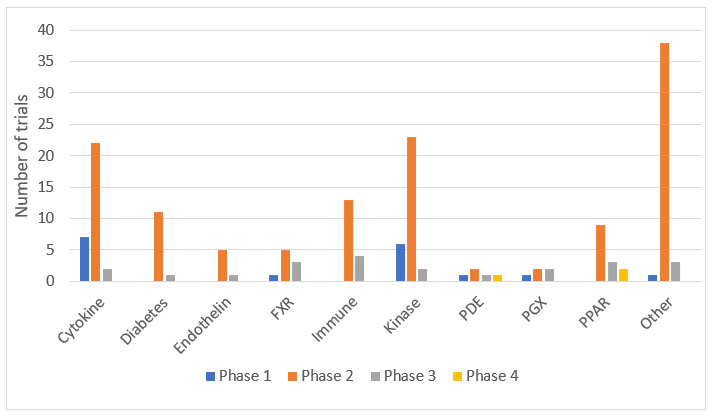
Out of 172 clinical trials we have identified, the highest proportion were in liver (65 [37.8%]) and lung (64 [37.2%]) fibrosis. The trials in heart, kidney and skin were relatively lower: there were 5 (2.9%) trials in heart, 14 (8.1%) in kidney and 24 (14%) in skin fibrosis. These results suggest that more candidate drugs are pursued in lung and liver fibrosis than skin, heart or kidney. There were 17 (10.1%) Phase 1, 130 (77.4%) Phase 2, 22 (13.1%) Phase 3 and 3 (1.8%) Phase 4 studies indicating a steep decrease from Phase 2 to Phase 3 studies. There were 130 Phase 2 and 22 Phase 3 trials suggesting an attrition rate of 83%. The distribution of each phase divided by organ is shown in , which suggests that while there is more activity in Phase 2 trials in liver fibrosis, when compared to the other organs, there seems to be more Phase 1 activity in lung fibrosis.
Figure 1. The distribution of clinical trials divided by organs and phases.
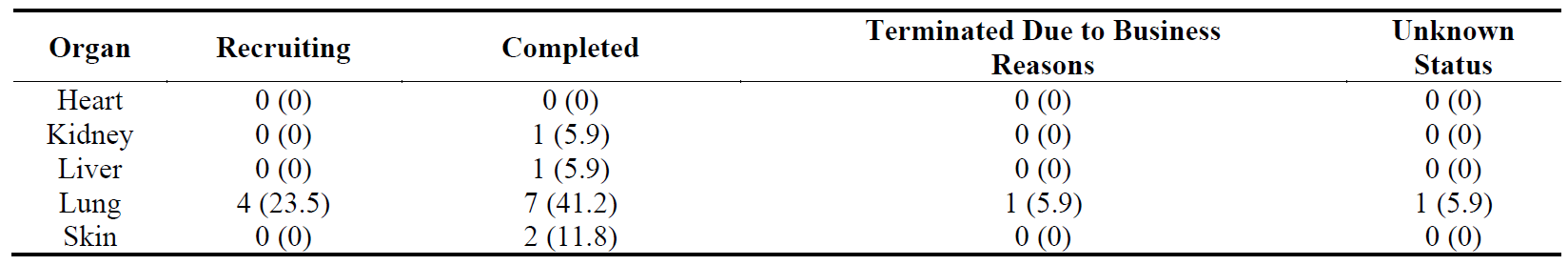
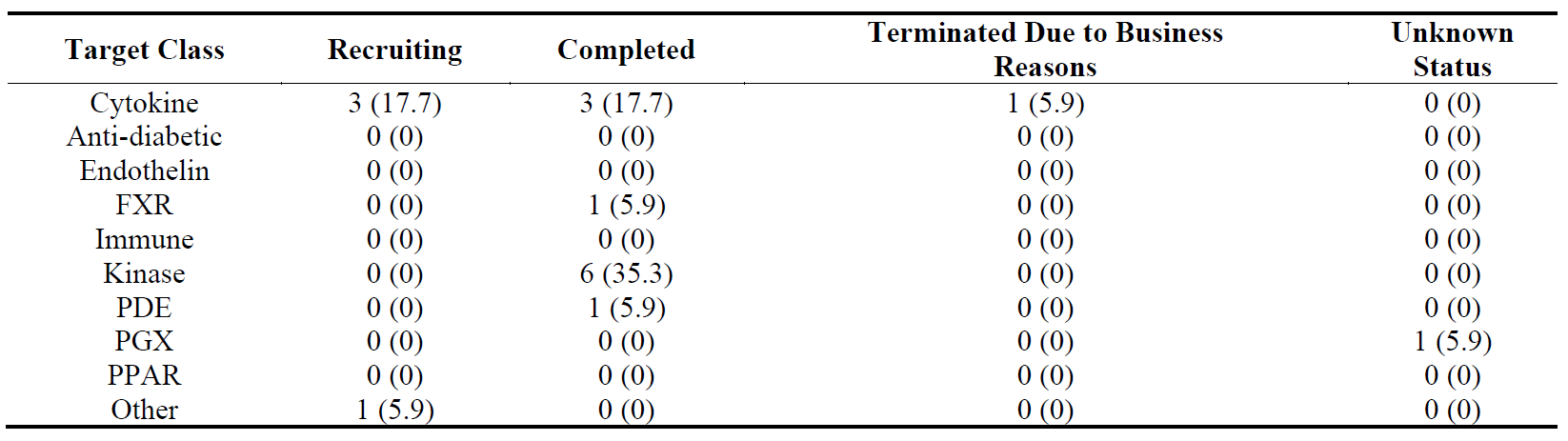
The distribution of the trials divided by organ and their progress are shown in , , and . The distribution pattern of 17 Phase 1 trials () correlates with , suggesting that lung fibrosis has more activity in Phase 1 than the other organs, but the number of recruiting Phase 1 trials are relatively low.
Table 12. Distribution of 17 Phase 1 clinical trials in five major fibrotic diseases (heart, kidney, liver, lung and skin) according to their stages in clinical development. The percentages are shown in brackets.
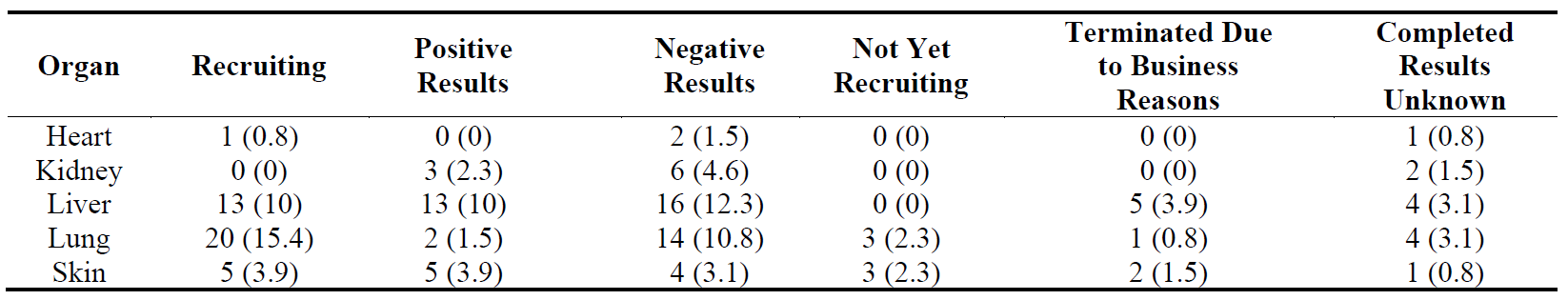
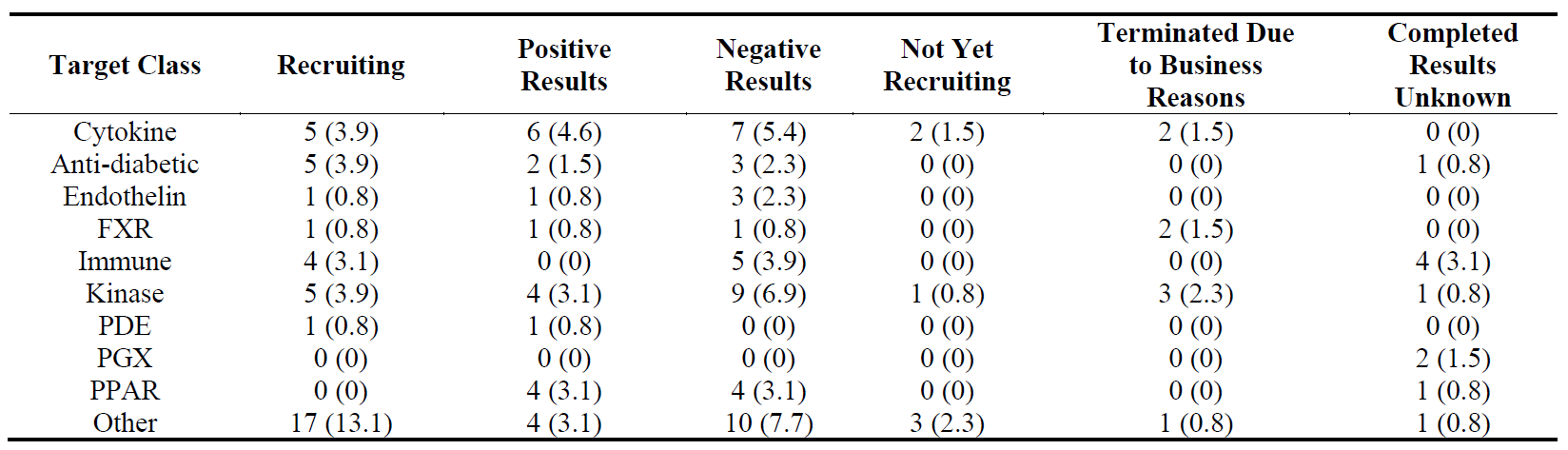
The distribution of 130 Phase 2 trials () shows that while there is more activity in liver fibrosis in this phase than the other organs (also shown in ), most of these trials either have negative results, are terminated due to business reasons or are completed but do not have results, which leaves only 13 trials with positive results and 13 recruiting trials. In contrast, lung fibrosis has 20 recruiting trials suggesting a high activity in this organ in Phase 2. There were 23 trials with positive results which would give an 18% success rate (82% attrition rate). If we compare the number of trials with positive results to those with negative results, only trials in skin fibrosis had more positive results than negative results.
Table 13. Distribution of 130 Phase 2 clinical trials in five major fibrotic diseases (heart, kidney, liver, lung and skin) according to their stages in clinical development. The percentages are shown in brackets.
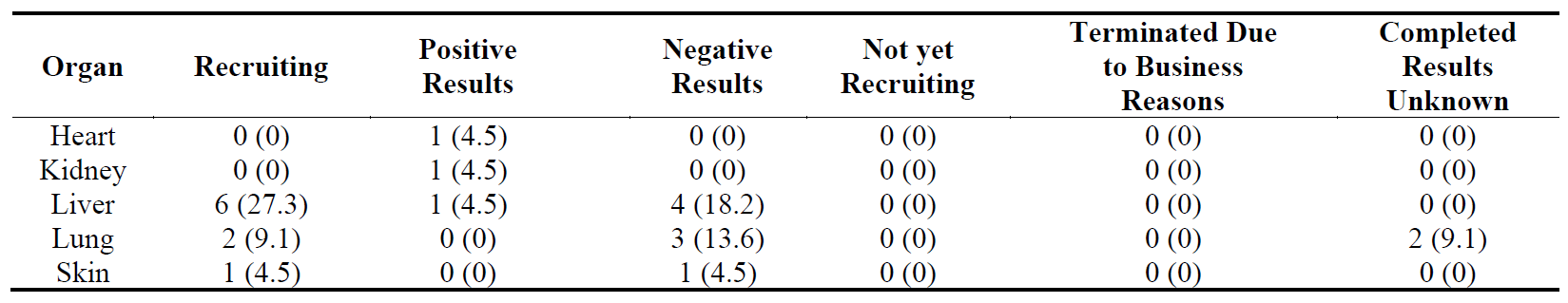
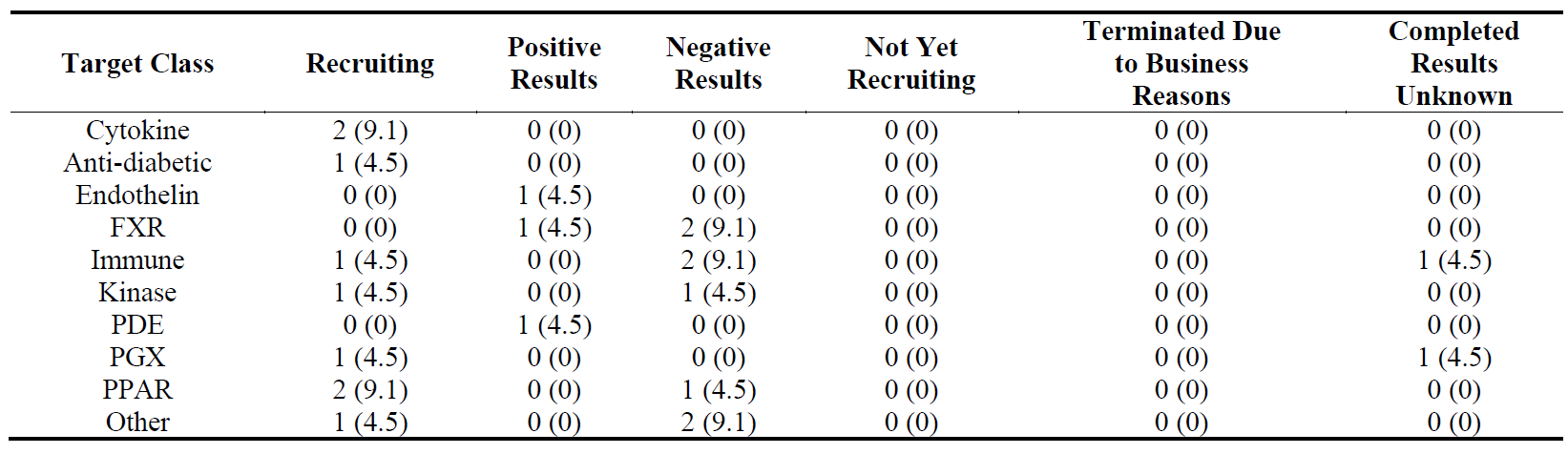
Out of 22 Phase 3 trials, the recruiting trials are in liver (6), lung (2) and skin (1) () suggesting a higher activity in liver fibrosis in Phase 3 than the other organs. Liver and lung fibrosis seem to have a similar number of trials with positive and negative results.
Table 14. Distribution of 21 Phase 3 clinical trials in five major fibrotic diseases (heart, kidney, liver, lung and skin) according to their stages in clinical development. The percentages are shown in brackets.
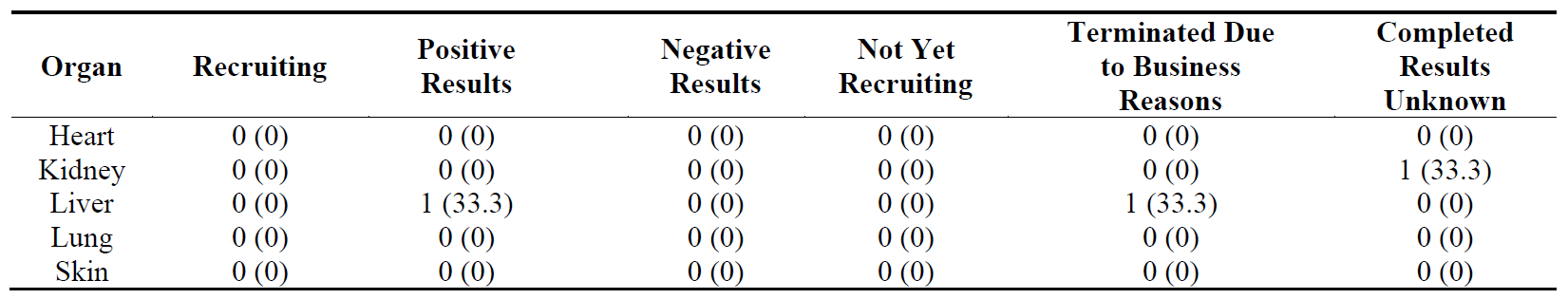
We have identified only 3 Phase 4 trials. The only one with positive results was lobeglitazone for liver fibrosis ( and ). One Phase 4 trial in liver fibrosis was terminated due to discontinuation of the drug (rosiglitazone, and ) and one Phase 4 study in kidney fibrosis was completed but the results have not been posted or published (pentoxifylline, and ).
Table 15. Distribution of 3 Phase 4 clinical trials in five major fibrotic diseases (heart, kidney, liver, lung and skin) according to their stages in clinical development. The percentages are shown in brackets.
5.2. Distribution According to Target Classes
When 172 trials were distributed according to their target classes, the highest number of trials were observed in cytokines (31 [18.5%]), kinases (31 [18.5%]) and other (42 [25%]) classes (). With most trials in Phase 2, the most common target classes observed were in cytokines, kinases, anti-diabetic drugs, immune and other classes ().
Figure 2. The distribution of clinical trials according to the target classes and the phases.
The distribution of 17 Phase 1 trials according to their target classes and progression is shown in . More than a third of completed Phase 1 trials were in “other” target class.
Table 16. Distribution of 17 Phase 1 clinical trials in ten target classes according to their stages in clinical development. The percentages are shown in brackets.
The distribution of 130 Phase 2 trials according to their target classes and progression is shown in . Although there were 17 trials recruiting in “other” target class, the number of trials with negative results (10) in this class was also high. The highest number of trials with positive results was in “cytokine” class. When we compare the numbers of trials with positive results to those with negative results, none of the target classes had more trials with positive results than those with negative results.
Table 17. Distribution of 130 Phase 2 clinical trials in ten target classes according to their stages in clinical development. The percentages are shown in brackets.
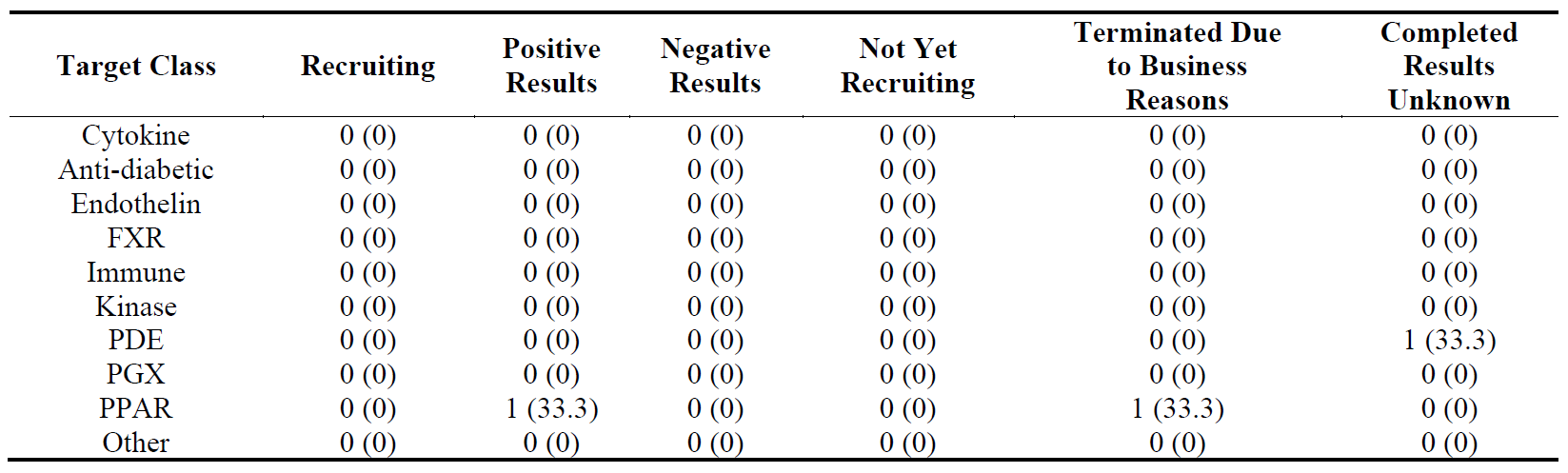
The distribution of 22 Phase 3 trials according to their targets and progression is shown in . The trials with negative results were observed in FXR, immune, kinase, PPAR and other classes.
Table 18. Distribution of 22 Phase 3 clinical trials in ten target classes according to their stages in clinical development. The percentages are shown in brackets.
The distribution of 3 Phase 4 trials according to their targets and progression is shown in . As mentioned above, a Phase 4 trial with the PDE-inhibitor pentoxifylline was completed but the results have not been posted. PPAR agonist lobeglitazone gave a postive result but rosiglitazone was terminated due to discontinuation of the drug.
Table 19. Distribution of 3 Phase 4 clinical trials in ten target classes according to their stages in clinical development. The percentages are shown in brackets.
6. Discussion
The search for novel anti-fibrotic drugs has been underway for decades. The anti-fibrotic drug discovery field has two major impetuses: (1) Fibrotic diseases can cause significant morbidity, mortality and burden on patients and healthcare systems, and the patient numbers are significant. The numbers are suggested to increase because of COVID-19 infections and increased prevalence of metabolic diseases. (2) The market size is significantly large: the anti-fibrotic drug market is estimated to have reached $30B per annum. Despite these significant demands from the markets, patients and the healthcare providers, the number of approved anti-fibrotic drugs are limited in number, safety and efficacy, suggesting a slow pace of drug discovery in this field. We therefore reviewed and analyzed the current landscape of drugs being developed in five major fibrotic diseases with the aim of finding and exploring trends and raising questions for the possible reasons for the slow pace. Although there have been previous reviews on anti-fibrotic drug development in individual fibrotic diseases such as lung [159], liver [160,161], kidney [162], heart [163] and skin [164], the reviews of all major fibrotic diseases have been limited [165,166,167]. To our knowledge this is the first study that compares the clinical trial activity in different diseases and target classes and analyses attrition rates.
The analysis of 172 clinical trials identified in this study revealed that the majority of the studies are currently focusing on lung and liver fibrosis with less focus on skin, heart and kidney. The numbers of clinical trials in liver fibrosis might be skewed because of drugs for metabolic diseases such as PPAR agonists and anti-diabetic drugs being included. Nevertheless, this trend may be the result of previous success, particularly in lung fibrosis, which may have given impetus and clinical trial experience to that particular field. It is envisaged that the recent success with resmetirom will give a similar boost to the liver fibrosis field.
Our analysis also revealed that there was a steep decline in the number of trials from Phase 2 (130) to Phase 3 (22) in all five fibrotic diseases. This suggests attrition of candidate molecules in Phase 2 trials at a rate of ~83%. Similarly, the proportion of Phase 2 studies with positive results is 18%, suggesting an 82% attrition rate. Such high rates are not unexpected since attrition rates as high as 70–80% in Phase 2 is common across all therapeutic areas, although there have been some improvements in recent years [168,169]. Whether an 83% attrition rate at Phase 2 is higher than the industry norm would require more in-depth analysis. However, we think it is safe to conclude that the fibrosis field does suffer from a high attrition rate in Phase 2.
When we analyzed the attrition rates according to the organs, the number of Phase 2 trials with positive results were higher than those with negative results, in skin fibrosis only. This suggests that the trend of high attrition may not be uniform across diseases. When the attrition rates were analyzed against the target classes, no target class had a higher number of trials with positive results than those with negative results, suggesting that a target class cannot be singled out with our analysis.
High attrition in Phase 2 has been attributed to lack of efficacy, poor safety and poor commercial viability [170]. In our analysis, several of the Phase 2 studies with negative results stated that the primary endpoint of the study was not reached, while the candidate molecule was well tolerated by the patients, suggesting that the main driver in Phase 2 attrition in fibrosis field is the lack of efficacy.
The lack of efficacy in Phase 2 trials in the fibrosis field is a known and recognized problem [171,172,173]. This has been partly attributed to the timing of the treatment. The pathology of fibrosis is unique as it gives only a narrow window of opportunity for intervention. During the inflammatory phase the repetitive injury triggers inflammation which activates pro-fibrotic elements that lead to transformation of fibroblasts and other cells to myofibroblasts. Once the myofibroblasts have terminally differentiated and the ECM proteins are produced and deposited, the tissue injury becomes irreversible [174,175,176]. In hypertrophic scars it is possible to know the time of the injury, allowing intervention with anti-fibrotic treatment before the ECMs are deposited, and this results in less scarring [177]. It has been suggested that early diagnosis and intervention with anti-fibrotic drugs pirfenidone and nintedanib may result in improving outcomes in IPF [178]. Similarly in liver fibrosis, since early diagnosis and intervention is known to deliver better outcomes, population screening has been suggested [179]. In heart fibrosis, early identification and treatment of patients with HFpEF has been suggested to be important for achieving optimal outcomes [180]. It would be interesting to see whether the efficacy of new anti-fibrotic drug resmetirom would differ between early- and late-stage liver disease. Additional evidence to support the notion that early intervention can prevent fibrosis comes from studies we have conducted in Peyronie’s disease (PD). PD is a fibrotic disease of the penis and can cause penile curvature, pain during erection and erectile dysfunction. The acute (inflammatory) phase of PD can last up to 6 months and usually presents with pain during erection. After the acute phase, pain subsides, and the chronic phase begins. Studies have shown that when treatment was given during the acute phase, before the fibrotic plaque was established, significant clinical improvement was observed [181]. For example, tamoxifen treatment or a combination of tamoxifen with a PDE5 inhibitor could prevent new fibrosis formation and negated the need for surgery [182,183,184]. When treatment was given during the chronic phase, however, there was no improvement [185]. These studies suggest that the design of the clinical trial, the correct patient inclusion and exclusion and timing of the treatment are of paramount importance.
To differentiate patients by early vs late fibrosis, clinical signs, symptoms and biomarkers that can differentiate the stages of fibrotic diseases are needed. As in the example of PD given above, the clinical symptom of pain, and a non-invasive easy way of measuring penile curvature, or penile ultrasound are ideal biomarkers for disease staging. However, this becomes more complicated in major fibrotic diseases as it becomes more difficult to diagnose fibrosis per se. For example, in liver fibrosis non-invasive clinical trial endpoints that are consistent for both Phase 2 and Phase 3 are still needed [172,186]. In cardiac fibrosis, again non-invasive biomarkers for fibrosis are needed, as endomyocardial biopsy, which is the gold standard for the diagnosis of myocardial fibrosis, has limitations in terms of clinical application [187].
Apart from study design and timing of the treatment, another factor that may be contributing to Phase 2 attrition is suggested to be the compensatory mechanisms in fibrosis. Fibrosis is fundamentally aberrant wound healing which has high levels of redundancy and compensatory mechanisms [188]. When a single target/pathway is inhibited, another pathway can take over/compensate for the loss of function. Therefore, one may need to target several pathways to achieve a meaningful functional response. This may be one of the reasons why we are seeing an increase in the combination of multiple drug candidates, in order to target different pathways, in the fibrosis field [189]. Another potential solution to this problem is the discovery of candidate molecules using a phenotypic approach rather than a target-based approach. Although historically older than the target-based approach, phenotypic drug discovery has recently re-emerged and has been shown to be more efficient in discovering first-in-class drugs than the target-based approach [190]. With the phenotypic approach, molecules are selected based on their action on a disease-associated phenotype rather than a single molecular target. This approach is target-agnostic and pathway-unbiased, making it a perfect alternative approach in the fibrosis field. There have been several compound screening campaigns using this approach for hypertrophic scars [191], lung [192], liver [193], kidney [194] & heart [195] fibrosis, and PD [196]. The drug combination (tamoxifen + PDE5 inhibitor) which was discovered through a phenotypic approach has been successfully translated into clinical outcomes in PD as mentioned above [182,183,184]. Further studies would be required to see whether other molecules that are identified using the phenotypic approach would find utility in the clinics.
Limitations
Despite our best efforts to capture all the clinical trials in five major fibrotic diseases, it is possible that we may have missed some. This should not count as ignoring or dismissing any study in the field. We recognize and acknowledge that the field is active, and trials may have started or concluded while this manuscript was being prepared.
7. Conclusions
Our analysis of the clinical trials in five major fibrotic diseases have revealed that the drug discovery and development activities in the field are focused on lung and liver, are slow paced and have high attrition in Phase 2. Better clinical trial design with better disease staging biomarkers and a phenotypic approach rather than a target-based approach are suggested to be potential solutions. To our knowledge, this is the first study that has estimated an 83% attrition rate in Phase 2 studies for major fibrotic diseases.
Author Contributions
Conceptualization, A.R.L. and S.C.; Methodology, A.R.L. and S.C.; Investigation and Data Curation, D.S., D.C.S. and S.C.; Writing—Original Draft Preparation, S.C.; Writing—Review & Editing, A.R.L., S.L.H., K.M.F., D.S., D.C.S., S.C.; Supervision, A.R.L.
Ethics Statement
Informed Consent Statement
Funding
This research received no external funding.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
1.
Wynn TA. Fibrotic disease and the TH1/TH2 paradigm.
Nat. Rev. Immunol. 2004,
4, 583–594. doi:10.1038/nri1412.
[Google Scholar]
2.
Parhizgar P, Yazdankhah N, Rzepka AM, Chung KYC, Ali I, Lai Fat Fur R, et al. Beyond Acute COVID-19: A Review of Long-term Cardiovascular Outcomes.
Can. J. Cardiol. 2023,
39, 726–740. doi:10.1016/j.cjca.2023.01.031.
[Google Scholar]
3.
Alrajhi NN. Post-COVID-19 pulmonary fibrosis: An ongoing concern.
Ann. Thorac. Med. 2023,
18, 173–181. doi:10.4103/atm.atm_7_23.
[Google Scholar]
4.
Senior M. Fighting fibrosis.
Nat. Biotechnol. 2022,
40, 1169–1173. doi:10.1038/s41587-022-01412-0.
[Google Scholar]
5.
Raghu G, Remy-Jardin M, Richeldi L, Thomson CC, Inoue Y, Johkoh T, et al. Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults: An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline.
Am. J. Respir. Crit. Care Med. 2022,
205, e18–e47. doi:10.1164/rccm.202202-0399ST.
[Google Scholar]
6.
Maher TM, Bendstrup E, Dron L, Langley J, Smith G, Khalid JM, et al. Global incidence and prevalence of idiopathic pulmonary fibrosis.
Respir. Res. 2021,
22, 197. doi:10.1186/s12931-021-01791-z.
[Google Scholar]
7.
Wilson MS, Wynn TA. Pulmonary fibrosis: Pathogenesis, etiology and regulation.
Mucosal Immunol. 2009,
2, 103–121. doi:10.1038/mi.2008.85.
[Google Scholar]
8.
Hunninghake GM, Quesada-Arias LD, Carmichael NE, Martinez Manzano JM, Poli De Frías S, Baumgartner MA, et al. Interstitial Lung Disease in Relatives of Patients with Pulmonary Fibrosis.
Am. J. Respir. Crit. Care Med. 2020,
201, 1240–1248. doi:10.1164/rccm.201908-1571OC.
[Google Scholar]
9.
Shah RM, Kolansky AM, Kligerman S. Thin-Section CT in the Categorization and Management of Pulmonary Fibrosis including Recently Defined Progressive Pulmonary Fibrosis.
Radiol. Cardiothorac. Imaging 2024,
6, e230135. doi:10.1148/ryct.230135.
[Google Scholar]
10.
Platenburg MGJP, van der Vis JJ, Grutters JC, van Moorsel CHM. Decreased Survival and Lung Function in Progressive Pulmonary Fibrosis.
Medicina 2023,
59, 296. doi:10.3390/medicina59020296.
[Google Scholar]
11.
Nathan SD, Shlobin OA, Weir N, Ahmad S, Kaldjob JM, Battle E, et al. Long-term Course and Prognosis of Idiopathic Pulmonary Fibrosis in the New Millennium.
Chest 2011,
140, 221–229. doi:10.1378/chest.10-2572.
[Google Scholar]
12.
Zheng Q, Cox IA, Campbell JA, Xia Q, Otahal P, de Graaff B, et al. Mortality and survival in idiopathic pulmonary fibrosis: A systematic review and meta-analysis.
ERJ Open Res. 2022,
8, 00591–02021. doi:10.1183/23120541.00591-2021.
[Google Scholar]
13.
Wuyts WA, Agostini C, Antoniou KM, Bouros D, Chambers RC, Cottin V, et al. The pathogenesis of pulmonary fibrosis: A moving target.
Eur. Respir. J. 2013,
41, 1207–1218. doi:10.1183/09031936.00073012.
[Google Scholar]
14.
Ortiz-Zapater E, Signes-Costa J, Montero P, Roger I. Lung Fibrosis and Fibrosis in the Lungs: Is It All about Myofibroblasts?
Biomedicines 2022,
10, 1423. doi:10.3390/biomedicines10061423.
[Google Scholar]
15.
Deng Z, Fear MW, Suk Choi Y, Wood FM, Allahham A, Mutsaers SE, et al. The extracellular matrix and mechanotransduction in pulmonary fibrosis.
Int. J. Biochem. Cell Biol. 2020,
126, 105802. doi:10.1016/j.biocel.2020.105802.
[Google Scholar]
16.
Burgstaller G, Oehrle B, Gerckens M, White ES, Schiller HB, Eickelberg O. The instructive extracellular matrix of the lung: Basic composition and alterations in chronic lung disease.
Eur. Respir. J. 2017,
50, 1601805. doi:10.1183/13993003.01805-2016.
[Google Scholar]
17.
Glass DS, Grossfeld D, Renna HA, Agarwala P, Spiegler P, DeLeon J, et al. Idiopathic pulmonary fibrosis: Current and future treatment.
Clin. Respir. J. 2022,
16, 84–96. doi:10.1111/crj.13466.
[Google Scholar]
18.
Maher TM, Wuyts W. Management of Fibrosing Interstitial Lung Diseases.
Adv. Ther. 2019,
36, 1518–1531. doi:10.1007/s12325-019-00992-9.
[Google Scholar]
19.
Pope JE, Denton CP, Johnson SR, Fernandez-Codina A, Hudson M, Nevskaya T. State-of-the-art evidence in the treatment of systemic sclerosis.
Nat. Rev. Rheumatol. 2023,
19, 212–226. doi:10.1038/s41584-023-00909-5.
[Google Scholar]
20.
Gourdie RG, Dimmeler S, Kohl P. Novel therapeutic strategies targeting fibroblasts and fibrosis in heart disease.
Nat. Rev. Drug Discov. 2016,
15, 620–638. doi:10.1038/nrd.2016.89.
[Google Scholar]
21.
Shah KS, Xu H, Matsouaka RA, Bhatt DL, Heidenreich PA, Hernandez AF, et al. Heart Failure With Preserved, Borderline, and Reduced Ejection Fraction.
J. Am. Coll. Cardiol. 2017,
70, 2476–2486. doi:10.1016/j.jacc.2017.08.074.
[Google Scholar]
22.
Teramoto K, Teng T-HK, Chandramouli C, Tromp J, Sakata Y, Lam CS. Epidemiology and Clinical Features of Heart Failure with Preserved Ejection Fraction.
Card. Fail. Rev. 2022,
8, e27. doi:10.15420/cfr.2022.06.
[Google Scholar]
23.
Graziani F, Varone F, Crea F, Richeldi L. Treating heart failure with preserved ejection fraction: Learning from pulmonary fibrosis.
Eur. J. Heart Fail. 2018,
20, 1385–1391. doi:10.1002/ejhf.1286.
[Google Scholar]
24.
Kong P, Christia P, Frangogiannis NG. The pathogenesis of cardiac fibrosis.
Cell. Mol. Life Sci. 2014,
71, 549–574. doi:10.1007/s00018-013-1349-6.
[Google Scholar]
25.
Iwanaga Y, Aoyama T, Kihara Y, Onozawa Y, Yoneda T, Sasayama S. Excessive activation of matrix metalloproteinases coincides with left ventricular remodeling during transition from hypertrophy to heart failure in hypertensive rats.
J. Am. Coll. Cardiol. 2002,
39, 1384–1391. doi:10.1016/S0735-1097(02)01756-4.
[Google Scholar]
26.
Khan R, Sheppard R. Fibrosis in heart disease: Understanding the role of transforming growth factor-β
1 in cardiomyopathy, valvular disease and arrhythmia.
Immunology 2006,
118, 10–24. doi:10.1111/j.1365-2567.2006.02336.x.
[Google Scholar]
27.
Devarbhavi H, Asrani SK, Arab JP, Nartey YA, Pose E, Kamath PS. Global burden of liver disease: 2023 update.
J. Hepatol. 2023,
79, 516–537. doi:10.1016/j.jhep.2023.03.017.
[Google Scholar]
28.
Stein E, Cruz-Lemini M, Altamirano J, Ndugga N, Couper D, Abraldes JG, et al. Heavy daily alcohol intake at the population level predicts the weight of alcohol in cirrhosis burden worldwide.
J. Hepatol. 2016,
65, 998–1005. doi:10.1016/j.jhep.2016.06.018.
[Google Scholar]
29.
Younossi Z, Anstee QM, Marietti M, Hardy T, Henry L, Eslam M, et al. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention.
Nat. Rev. Gastroenterol. Hepatol. 2018,
15, 11–20. doi:10.1038/nrgastro.2017.109.
[Google Scholar]
30.
Teng ML, Ng CH, Huang DQ, Chan KE, Tan DJ, Lim WH, et al. Global incidence and prevalence of nonalcoholic fatty liver disease.
Clin. Mol. Hepatol. 2023,
29, S32–S42. doi:10.3350/cmh.2022.0365.
[Google Scholar]
31.
Rippe RA. Liver fibrosis: signals leading to the amplification of the fibrogenic hepatic stellate cell.
Front. Biosci. 2003,
8, 887. doi:10.2741/887.
[Google Scholar]
32.
Friedman SL, Roll FJ, Boyles J, Bissell DM. Hepatic lipocytes: The principal collagen-producing cells of normal rat liver.
Proc. Natl. Acad. Sci. USA 1985,
82, 8681–8685. doi:10.1073/pnas.82.24.8681.
[Google Scholar]
33.
Magness ST, Bataller R, Yang L, Brenner DA. A dual reporter gene transgenic mouse demonstrates heterogeneity in hepatic fibrogenic cell populations.
Hepatology 2004,
40, 1151–1159. doi:10.1002/hep.20427.
[Google Scholar]
34.
Kinnman N. Peribiliary myofibroblasts in biliary type liver fibrosis.
Front. Biosci. 2002,
7, A790. doi:10.2741/A790.
[Google Scholar]
35.
Bataller R, Brenner DA. Liver fibrosis.
J. Clin. Investig. 2005,
115, 209–218. doi:10.1172/JCI24282.
[Google Scholar]
36.
Dufour J-F, Anstee QM, Bugianesi E, Harrison S, Loomba R, Paradis V, et al. Current therapies and new developments in NASH.
Gut 2022,
71, 2123–2134. doi:10.1136/gutjnl-2021-326874.
[Google Scholar]
37.
Heidelbaugh JJ, Sherbondy M. Cirrhosis and chronic liver failure: Part II. Complications and treatment.
Am. Fam. Physician 2006,
74, 767–776.
[Google Scholar]
38.
Webster AC, Nagler EV, Morton RL, Masson P. Chronic Kidney Disease.
Lancet 2017,
389, 1238–1252. doi:10.1016/S0140-6736(16)32064-5.
[Google Scholar]
39.
Kovesdy CP. Epidemiology of chronic kidney disease: An update 2022.
Kidney Int. Suppl. 2022,
12, 7–11. doi:10.1016/j.kisu.2021.11.003.
[Google Scholar]
40.
Feng X, Hou N, Chen Z, Liu J, Li X, Sun X, et al. Secular trends of epidemiologic patterns of chronic kidney disease over three decades: An updated analysis of the Global Burden of Disease Study 2019.
BMJ Open 2023,
13, e064540. doi:10.1136/bmjopen-2022-064540.
[Google Scholar]
41.
Huang R, Fu P, Ma L. Kidney fibrosis: From mechanisms to therapeutic medicines. Signal Transduct.
Target. Ther. 2023,
8, 129. doi:10.1038/s41392-023-01379-7.
[Google Scholar]
42.
Li L, Fu H, Liu Y. The fibrogenic niche in kidney fibrosis: Components and mechanisms.
Nat. Rev. Nephrol. 2022,
18, 545–557. doi:10.1038/s41581-022-00590-z.
[Google Scholar]
43.
Afsar B, Afsar RE, Dagel T, Kaya E, Erus S, Ortiz A, et al. Capillary rarefaction from the kidney point of view.
Clin. Kidney J. 2018,
11, 295–301. doi:10.1093/ckj/sfx133.
[Google Scholar]
44.
Hou FF, Liu Y. New insights into the pathogenesis and therapeutics of kidney fibrosis.
Kidney Int. Suppl. 2014,
4, 1. doi:10.1038/kisup.2014.1.
[Google Scholar]
45.
Elendu C, Elendu RC, Enyong JM, Ibhiedu JO, Ishola IV, Egbunu EO, et al. Comprehensive review of current management guidelines of chronic kidney disease.
Medicine 2023,
102, e33984. doi:10.1097/MD.0000000000033984.
[Google Scholar]
46.
Morgan ND, Hummers LK. Scleroderma Mimickers.
Curr. Treat. Opt. Rheumatol. 2016,
2, 69–84. doi:10.1007/s40674-016-0038-7.
[Google Scholar]
47.
Cutolo M, Damjanov N, Ruaro B, Zekovic A, Smith V. Imaging of connective tissue diseases: Beyond visceral organ imaging?
Best. Pract. Res. Clin. Rheumatol. 2016,
30, 670–687. doi:10.1016/j.berh.2016.10.002.
[Google Scholar]
48.
Santiago T, Santiago M, Ruaro B, Salvador MJ, Cutolo M, da Silva JAP. Ultrasonography for the Assessment of Skin in Systemic Sclerosis: A Systematic Review.
Arthritis Care Res. 2019,
71, 563–574. doi:10.1002/acr.23597.
[Google Scholar]
49.
Careta MF, Romiti R. Localized scleroderma: Clinical spectrum and therapeutic update.
An. Bras. Dermatol. 2015,
90, 62–73. doi:10.1590/abd1806-4841.20152890.
[Google Scholar]
50.
Distler O, Cozzio A. Systemic sclerosis and localized scleroderma--current concepts and novel targets for therapy.
Semin. Immunopathol. 2016,
38, 87–95. doi:10.1007/s00281-015-0551-z.
[Google Scholar]
51.
Krieg T, Takehara K. Skin disease: A cardinal feature of systemic sclerosis.
Rheumatology 2009,
48 (Suppl 3), iii14–iii18. doi:10.1093/rheumatology/kep108.
[Google Scholar]
52.
Tian J, Kang S, Zhang D, Huang Y, Zhao M, Gui X, et al. Global, regional, and national incidence and prevalence of systemic sclerosis.
Clin. Immunol. 2023,
248, 109267. doi:10.1016/j.clim.2023.109267.
[Google Scholar]
53.
Asano Y. The Pathogenesis of Systemic Sclerosis: An Understanding Based on a Common Pathologic Cascade across Multiple Organs and Additional Organ-Specific Pathologies.
J. Clin. Med. 2020,
9, 2687. doi:10.3390/jcm9092687.
[Google Scholar]
54.
Tyndall AJ, Bannert B, Vonk M, Airo P, Cozzi F, Carreira PE, et al. Causes and risk factors for death in systemic sclerosis: A study from the EULAR Scleroderma Trials and Research (EUSTAR) database.
Ann. Rheum. Dis. 2010,
69, 1809–1815. doi:10.1136/ard.2009.114264.
[Google Scholar]
55.
Katsiari CG, Simopoulou T, Alexiou I, Sakkas LI. Immunotherapy of systemic sclerosis.
Hum Vaccin Immunother. 2018,
14, 2559–2567. doi:10.1080/21645515.2018.1491508.
[Google Scholar]
56.
Gauglitz GG, Korting HC, Pavicic T, Ruzicka T, Jeschke MG. Hypertrophic Scarring and Keloids: Pathomechanisms and Current and Emerging Treatment Strategies.
Mol. Med. 2011,
17, 113–125. doi:10.2119/molmed.2009.00153.
[Google Scholar]
57.
Mony MP, Harmon KA, Hess R, Dorafshar AH, Shafikhani SH. An Updated Review of Hypertrophic Scarring.
Cells 2023,
12, 678. doi:10.3390/cells12050678.
[Google Scholar]
58.
Basson R, Bayat A. Skin scarring: Latest update on objective assessment and optimal management.
Front. Med. 2022,
9, 942756. doi:10.3389/fmed.2022.942756.
[Google Scholar]
59.
Lian N, Li T. Growth factor pathways in hypertrophic scars: Molecular pathogenesis and therapeutic implications.
Biomed. Pharmacother. 2016,
84, 42–50. doi:10.1016/j.biopha.2016.09.010.
[Google Scholar]
60.
Shah PV, Balani P, Lopez AR, Nobleza CMN, Siddiqui M, Khan S. A Review of Pirfenidone as an Anti-Fibrotic in Idiopathic Pulmonary Fibrosis and Its Probable Role in Other Diseases. Cureus 2021, 13. doi:10.7759/cureus.12482.
61.
Schaefer CJ, Ruhrmund DW, Pan L, Seiwert SD, Kossen K. Antifibrotic activities of pirfenidone in animal models.
Eur. Respir. Rev. 2011,
20, 85–97. doi:10.1183/09059180.00001111.
[Google Scholar]
62.
Zang C, Zheng Y, Wang Y, Li L. The effects and safety of pirfenidone in the treatment of idiopathic pulmonary fibrosis: A meta-analysis and systematic review.
Eur. J. Med. Res. 2021,
26, 129. doi:10.1186/s40001-021-00601-y.
[Google Scholar]
63.
Cottin V, Maher T. Long-term clinical and real-world experience with pirfenidone in the treatment of idiopathic pulmonary fibrosis.
Eur. Respir. Rev. 2015,
24, 58–64. doi:10.1183/09059180.00011514.
[Google Scholar]
64.
Hayton C, Chaudhuri N. Managing Idiopathic Pulmonary Fibrosis: Which Drug for Which Patient?
Drugs Aging 2017,
34, 647–653. doi:10.1007/s40266-017-0488-0.
[Google Scholar]
65.
Roth GJ, Binder R, Colbatzky F, Dallinger C, Schlenker-Herceg R, Hilberg F, et al. Nintedanib: From Discovery to the Clinic.
J. Med. Chem. 2015,
58, 1053–1063. doi:10.1021/jm501562a.
[Google Scholar]
66.
Wollin L, Wex E, Pautsch A, Schnapp G, Hostettler KE, Stowasser S, et al. Mode of action of nintedanib in the treatment of idiopathic pulmonary fibrosis.
Eur. Respir. J. 2015,
45, 1434–1445. doi:10.1183/09031936.00174914.
[Google Scholar]
67.
Bonella F, Cottin V, Valenzuela C, Wijsenbeek M, Voss F, Rohr KB, et al. Meta-Analysis of Effect of Nintedanib on Reducing FVC Decline Across Interstitial Lung Diseases.
Adv. Ther. 2022,
39, 3392–3402. doi:10.1007/s12325-022-02145-x.
[Google Scholar]
68.
Wuyts WA, Maher TM, Wijsenbeek M, Koschel D, Martinez FJ, Stansen W, et al. Meta-analysis of effect of nintedanib on mortality in subjects with idiopathic pulmonary fibrosis (IPF) and other forms of progressive pulmonary fibrosis (PPF). In Idiopathic Interstitial Pneumonias; European Respiratory Society: Lausanne, Switzerland, 2023; p. PA2875. doi:10.1183/13993003.congress-2023.PA2875.
69.
Chen C-H, Lin H-C, Wang Y-H, Wang C-Y, Lin YS, Lai C-C. The safety of nintedanib for the treatment of interstitial lung disease: A systematic review and meta-analysis of randomized controlled trials.
PLoS ONE 2021,
16, e0251636. doi:10.1371/journal.pone.0251636.
[Google Scholar]
70.
Liu F, Wang L, Qi H, Wang J, Wang Y, Jiang W, et al. Nintedanib, a triple tyrosine kinase inhibitor, attenuates renal fibrosis in chronic kidney disease.
Clin. Sci. 2017,
131, 2125–2143. doi:10.1042/CS20170134.
[Google Scholar]
71.
Wollin L, Togbe D, Ryffel B. Effects of Nintedanib in an Animal Model of Liver Fibrosis.
Biomed. Res. Int. 2020,
2020, 3867198. doi:10.1155/2020/3867198.
[Google Scholar]
72.
Lin S, Huang S, Deng Z, Zhang Y, Huang L, Wu Y, et al. Discovery of a novel, liver-targeted thyroid hormone receptor-β agonist, CS271011, in the treatment of lipid metabolism disorders.
Front. Endocrinol. 2023,
14, 1109615. doi:10.3389/fendo.2023.1109615.
[Google Scholar]
73.
Kannt A, Wohlfart P, Madsen AN, Veidal SS, Feigh M, Schmoll D. Activation of thyroid hormone receptor-β improved disease activity and metabolism independent of body weight in a mouse model of non-alcoholic steatohepatitis and fibrosis.
Br. J. Pharmacol. 2021,
178, 2412–2423. doi:10.1111/bph.15427.
[Google Scholar]
74.
Harrison SA, Bashir MR, Guy CD, Zhou R, Moylan CA, Frias JP, et al. Resmetirom (MGL-3196) for the treatment of non-alcoholic steatohepatitis: A multicentre, randomised, double-blind, placebo-controlled, phase 2 trial.
Lancet 2019,
394, 2012–2024. doi:10.1016/S0140-6736(19)32517-6.
[Google Scholar]
75.
Harrison SA, Bedossa P, Guy CD, Schattenberg JM, Loomba R, Taub R, et al. A Phase 3, Randomized, Controlled Trial of Resmetirom in NASH with Liver Fibrosis.
New Engl. J. Med. 2024,
390, 497–509. doi:10.1056/NEJMoa2309000.
[Google Scholar]
76.
Harrison SA, Taub R, Neff GW, Lucas KJ, Labriola D, Moussa SE, et al. Resmetirom for nonalcoholic fatty liver disease: A randomized, double-blind, placebo-controlled phase 3 trial.
Nat. Med. 2023,
29, 2919–2928. doi:10.1038/s41591-023-02603-1.
[Google Scholar]
77.
Frangogiannis NG. Transforming growth factor–β in tissue fibrosis.
J. Exp. Med. 2020,
217, e20190103. doi:10.1084/jem.20190103.
[Google Scholar]
78.
Lafyatis R. Transforming growth factor β—At the centre of systemic sclerosis.
Nat. Rev. Rheumatol. 2014,
10, 706–719. doi:10.1038/nrrheum.2014.137.
[Google Scholar]
79.
Lipson KE, Wong C, Teng Y, Spong S. CTGF is a central mediator of tissue remodeling and fibrosis and its inhibition can reverse the process of fibrosis.
Fibrogenes. Tissue Repair. 2012,
5, S24. doi:10.1186/1755-1536-5-S1-S24.
[Google Scholar]
80.
Guzy RD, Bollenbecker S, Krick S. From the Gut to the Lung: Evidence of Antifibrotic Activity of Endocrine Fibroblast Growth Factor 19.
Am. J. Respir. Cell Mol. Biol. 2022,
67, 139–141. doi:10.1165/rcmb.2022-0057ED.
[Google Scholar]
81.
Jia M-Q, Guan C-X, Tao J-H, Zhou Y. Research Progress of Fibroblast Growth Factor 21 in Fibrotic Diseases.
Oxid. Med. Cell Longev. 2022,
2022, 5042762. doi:10.1155/2022/5042762.
[Google Scholar]
82.
Meng F, Khoso MH, Kang K, He Q, Cao Y, Jiang X, et al. FGF21 ameliorates hepatic fibrosis by multiple mechanisms.
Mol. Biol. Rep. 2021,
48, 7153–7163. doi:10.1007/s11033-021-06707-0.
[Google Scholar]
83.
Chui ZSW, Shen Q, Xu A. Current status and future perspectives of FGF21 analogues in clinical trials.
Trends Endocrinol. Metab. 2024,
35, 371–384. doi:10.1016/j.tem.2024.02.001.
[Google Scholar]
84.
Rinella ME, Lieu HD, Kowdley KV, Goodman ZD, Alkhouri N, Lawitz E, et al. A randomized, double-blind, placebo-controlled trial of aldafermin in patients with NASH and compensated cirrhosis.
Hepatology 2024,
79, 674–689. doi:10.1097/HEP.0000000000000607.
[Google Scholar]
85.
Wuyts WA, Valenzuela C, Jenkins G, Goldin JG, Kim GHJ, Jurek M, et al. Late Breaking Abstract—Safety, tolerability and antifibrotic activity of bexotegrast: Phase 2a INTEGRIS-IPF study (NCT04396756). In Idiopathic Interstitial Pneumonias; European Respiratory Society: Lausanne, Switzerland, 2023; p. OA1423. doi:10.1183/13993003.congress-2023.OA1423.
86.
Gale JD, Jensen J, Berman G, Freimuth W, Li G, Pleil A, et al. A Placebo-controlled Study of PF-06473871 (Anti-Connective Tissue Growth Factor Antisense Oligonucleotide) in Reducing Hypertrophic Skin Scarring.
Plast. Reconstr. Surg. Glob. Open 2018,
6, e1861. doi:10.1097/GOX.0000000000001861.
[Google Scholar]
87.
Vincenti F, Fervenza FC, Campbell KN, Diaz M, Gesualdo L, Nelson P, et al. A Phase 2, Double-Blind, Placebo-Controlled, Randomized Study of Fresolimumab in Patients with Steroid-Resistant Primary Focal Segmental Glomerulosclerosis.
Kidney Int. Rep. 2017,
2, 800–810. doi:10.1016/j.ekir.2017.03.011.
[Google Scholar]
88.
Rice LM, Padilla CM, McLaughlin SR, Mathes A, Ziemek J, Goummih S, et al. Fresolimumab treatment decreases biomarkers and improves clinical symptoms in systemic sclerosis patients.
J. Clin. Investig. 2015,
125, 2795–2807. doi:10.1172/JCI77958.
[Google Scholar]
89.
Abdelmalek MF, Sanyal AJ, Nakajima A, Neuschwander-Tetri BA, Goodman ZD, Lawitz EJ, et al. Pegbelfermin in Patients With Nonalcoholic Steatohepatitis and Compensated Cirrhosis (FALCON 2): A Randomized Phase 2b Study.
Clin. Gastroenterol. Hepatol. 2024,
22, 113–123.e9. doi:10.1016/j.cgh.2023.04.012.
[Google Scholar]
90.
Denton CP, Merkel PA, Furst DE, Khanna D, Emery P, Hsu VM, et al. Recombinant human anti–transforming growth factor β1 antibody therapy in systemic sclerosis: A multicenter, randomized, placebo-controlled phase I/II trial of CAT-192.
Arthritis Rheum. 2007,
56, 323–333. doi:10.1002/art.22289.
[Google Scholar]
91.
Voelker J, Berg PH, Sheetz M, Duffin K, Shen T, Moser B, et al. Anti–TGF-β1 Antibody Therapy in Patients with Diabetic Nephropathy.
J. Am. Soc. Nephrol. 2017,
28, 953–962. doi:10.1681/ASN.2015111230.
[Google Scholar]
92.
Kim S, Lim JH, Woo C-H. Therapeutic potential of targeting kinase inhibition in patients with idiopathic pulmonary fibrosis.
Yeungnam Univ. J. Med. 2020,
37, 269–276. doi:10.12701/yujm.2020.00458.
[Google Scholar]
93.
Cohen P, Cross D, Jänne PA. Kinase drug discovery 20 years after imatinib: Progress and future directions.
Nat. Rev. Drug Discov. 2021,
20, 551–569. doi:10.1038/s41573-021-00195-4.
[Google Scholar]
94.
Cai X, Liu X, Xie W, Ma A, Tan Y, Shang J, et al. Hydronidone for the Treatment of Liver Fibrosis Related to Chronic Hepatitis B: A Phase 2 Randomized Controlled Trial.
Clin. Gastroenterol. Hepatol. 2023,
21, 1893–1901.e7. doi:10.1016/j.cgh.2022.05.056.
[Google Scholar]
95.
Gordon J, Spiera R. Imatinib and the Treatment of Fibrosis: Recent Trials and Tribulations.
Curr. Rheumatol. Rep. 2011,
13, 51–58. doi:10.1007/s11926-010-0146-6.
[Google Scholar]
96.
Qiu YY, Zhang J, Zeng FY, Zhu YZ. Roles of the peroxisome proliferator-activated receptors (PPARs) in the pathogenesis of nonalcoholic fatty liver disease (NAFLD).
Pharmacol. Res. 2023,
192, 106786. doi:10.1016/j.phrs.2023.106786.
[Google Scholar]
97.
Lee Y, Kim JH, Kim SR, Jin HY, Rhee E-J, Cho YM, et al. Lobeglitazone, a Novel Thiazolidinedione, Improves Non-Alcoholic Fatty Liver Disease in Type 2 Diabetes: Its Efficacy and Predictive Factors Related to Responsiveness.
J. Korean Med. Sci. 2017,
32, 60. doi:10.3346/jkms.2017.32.1.60.
[Google Scholar]
98.
Nakajima A, Eguchi Y, Yoneda M, Imajo K, Tamaki N, Suganami H, et al. Randomised clinical trial: Pemafibrate, a novel selective peroxisome proliferator-activated receptor α modulator (SPPARMα), versus placebo in patients with non-alcoholic fatty liver disease.
Aliment. Pharmacol. Ther. 2021,
54, 1263–1277. doi:10.1111/apt.16596.
[Google Scholar]
99.
Francque SM, Bedossa P, Ratziu V, Anstee QM, Bugianesi E, Sanyal AJ, et al. A Randomized, Controlled Trial of the Pan-PPAR Agonist Lanifibranor in NASH.
New Engl. J. Med. 2021,
385, 1547–1558. doi:10.1056/NEJMoa2036205.
[Google Scholar]
100.
Sven MF, Pierre B, Manal FA, Quentin MA, Elisabetta B, Vlad R, et al. A randomised, double-blind, placebo-controlled, multi-centre, dose-range, proof-of-concept, 24-week treatment study of lanifibranor in adult subjects with non-alcoholic steatohepatitis: Design of the NATIVE study.
Contemp. Clin. Trials 2020,
98, 106170. doi:10.1016/j.cct.2020.106170.
[Google Scholar]
101.
Kamata S, Honda A, Ishii I. Current Clinical Trial Status and Future Prospects of PPAR-Targeted Drugs for Treating Nonalcoholic Fatty Liver Disease.
Biomolecules 2023,
13, 1264. doi:10.3390/biom13081264.
[Google Scholar]
102.
Ratziu V, Harrison SA, Francque S, Bedossa P, Lehert P, Serfaty L, et al. Elafibranor, an Agonist of the Peroxisome Proliferator−Activated Receptor−α and −δ, Induces Resolution of Nonalcoholic Steatohepatitis Without Fibrosis Worsening.
Gastroenterology 2016,
150, 1147–1159.e5. doi:10.1053/j.gastro.2016.01.038.
[Google Scholar]
103.
Maguire JJ, Davenport AP. Endothelin Receptors and Their Antagonists.
Semin. Nephrol. 2015,
35, 125–136. doi:10.1016/j.semnephrol.2015.02.002.
[Google Scholar]
104.
Clozel M, Salloukh H. Role of endothelin in fibrosis and anti-fibrotic potential of bosentan.
Ann. Med. 2005,
37, 2–12. doi:10.1080/07853890410018925.
[Google Scholar]
105.
Li S, Pan Y, Xin W, Yan C. The potential benefit of endothelin receptor antagonists’ therapy in idiopathic pulmonary fibrosis: A meta-analysis of results from randomized controlled trials.
Medicine 2022,
101, e29981. doi:10.1097/MD.0000000000029981.
[Google Scholar]
106.
Heerspink HJL, Parving H-H, Andress DL, Bakris G, Correa-Rotter R, Hou F-F, et al. Atrasentan and renal events in patients with type 2 diabetes and chronic kidney disease (SONAR): A double-blind, randomised, placebo-controlled trial.
Lancet 2019,
393, 1937–1947. doi:10.1016/S0140-6736(19)30772-X.
[Google Scholar]
107.
Heerspink HJL, Kiyosue A, Wheeler DC, Lin M, Wijkmark E, Carlson G, et al. Zibotentan in combination with dapagliflozin compared with dapagliflozin in patients with chronic kidney disease (ZENITH-CKD): A multicentre, randomised, active-controlled, phase 2b, clinical trial.
Lancet 2023,
402, 2004–2017. doi:10.1016/S0140-6736(23)02230-4.
[Google Scholar]
108.
Raghu G. Treatment of Idiopathic Pulmonary Fibrosis with Ambrisentan.
Ann. Intern. Med. 2013,
158, 641. doi:10.7326/0003-4819-158-9-201305070-00003.
[Google Scholar]
109.
King TE, Brown KK, Raghu G, du Bois RM, Lynch DA, Martinez F, et al. BUILD-3: A Randomized, Controlled Trial of Bosentan in Idiopathic Pulmonary Fibrosis.
Am. J. Respir. Crit. Care Med. 2011,
184, 92–99. doi:10.1164/rccm.201011-1874OC.
[Google Scholar]
110.
Raghu G, Million-Rousseau R, Morganti A, Perchenet L, Behr J. Macitentan for the treatment of idiopathic pulmonary fibrosis: The randomised controlled MUSIC trial.
Eur. Respir. J. 2013,
42, 1622–1632. doi:10.1183/09031936.00104612.
[Google Scholar]
111.
Schinner E, Wetzl V, Schramm A, Kees F, Sandner P, Stasch J, et al. Inhibition of the TGF β signalling pathway by cGMP and cGMP-dependent kinase I in renal fibrosis.
FEBS Open Bio. 2017,
7, 550–561. doi:10.1002/2211-5463.12202.
[Google Scholar]
112.
Insel PA, Murray F, Yokoyama U, Romano S, Yun H, Brown L, et al. cAMP and Epac in the regulation of tissue fibrosis.
Br. J. Pharmacol. 2012,
166, 447–456. doi:10.1111/j.1476-5381.2012.01847.x.
[Google Scholar]
113.
Guazzi M, Vicenzi M, Arena R, Guazzi MD. PDE5 Inhibition With Sildenafil Improves Left Ventricular Diastolic Function, Cardiac Geometry, and Clinical Status in Patients With Stable Systolic Heart Failure.
Circ. Heart Fail. 2011,
4, 8–17. doi:10.1161/CIRCHEARTFAILURE.110.944694.
[Google Scholar]
114.
Richeldi L, Azuma A, Cottin V, Hesslinger C, Stowasser S, Valenzuela C, et al. Trial of a Preferential Phosphodiesterase 4B Inhibitor for Idiopathic Pulmonary Fibrosis.
New Engl. J. Med. 2022,
386, 2178–2187. doi:10.1056/NEJMoa2201737.
[Google Scholar]
115.
Hu Y, Li H, Zhang H, Chen X, Chen J, Xu Z, et al. ZSP1601, a novel pan-phosphodiesterase inhibitor for the treatment of NAFLD, A randomized, placebo-controlled phase Ib/IIa trial.
Nat. Commun. 2023,
14, 6409. doi:10.1038/s41467-023-42162-0.
[Google Scholar]
116.
Sabounjian L, Graham P, Wu L, Braman V, Cheng C, Liu J, et al. A First-in-Patient, Multicenter, Double-Blind, 2-Arm, Placebo-Controlled, Randomized Safety and Tolerability Study of a Novel Oral Drug Candidate, CTP-499, in Chronic Kidney Disease.
Clin. Pharmacol. Drug Dev. 2016,
5, 314–325. doi:10.1002/cpdd.241.
[Google Scholar]
117.
Wick G, Backovic A, Rabensteiner E, Plank N, Schwentner C, Sgonc R. The immunology of fibrosis: Innate and adaptive responses.
Trends Immunol. 2010,
31, 110–119. doi:10.1016/j.it.2009.12.001.
[Google Scholar]
118.
Khanna D, Lin CJF, Furst DE, Goldin J, Kim G, Kuwana M, et al. Tocilizumab in systemic sclerosis: A randomised, double-blind, placebo-controlled, phase 3 trial.
Lancet Respir. Med. 2020,
8, 963–974. doi:10.1016/S2213-2600(20)30318-0.
[Google Scholar]
119.
Adorini L, Trauner M. FXR agonists in NASH treatment.
J. Hepatol. 2023,
79, 1317–1331. doi:10.1016/j.jhep.2023.07.034.
[Google Scholar]
120.
Ratziu V, Harrison SA, Loustaud-Ratti V, Bureau C, Lawitz E, Abdelmalek M, et al. Hepatic and renal improvements with FXR agonist vonafexor in individuals with suspected fibrotic NASH.
J. Hepatol. 2023,
78, 479–492. doi:10.1016/j.jhep.2022.10.023.
[Google Scholar]
121.
Ricciotti E, FitzGerald GA. Prostaglandins and Inflammation.
Arterioscler. Thromb. Vasc. Biol. 2011,
31, 986–1000. doi:10.1161/ATVBAHA.110.207449.
[Google Scholar]
122.
Nasrallah R, Hébert RL. Prostacyclin signaling in the kidney: Implications for health and disease.
Am. J. Physiol. -Ren. Physiol. 2005,
289, F235–F246. doi:10.1152/ajprenal.00454.2004.
[Google Scholar]
123.
Zhu Y, Liu Y, Zhou W, Xiang R, Jiang L, Huang K, et al. A prostacyclin analogue, iloprost, protects from bleomycin-induced pulmonary fibrosis in mice.
Respir. Res. 2010,
11, 34. doi:10.1186/1465-9921-11-34.
[Google Scholar]
124.
Song W-L, Ricciotti E, Liang X, Grosser T, Grant GR, FitzGerald GA. Lipocalin-Like Prostaglandin D Synthase but Not Hemopoietic Prostaglandin D Synthase Deletion Causes Hypertension and Accelerates Thrombogenesis in Mice.
J. Pharmacol. Exp. Ther. 2018,
367, 425–432. doi:10.1124/jpet.118.250936.
[Google Scholar]
125.
Rittchen S, Heinemann A. Therapeutic Potential of Hematopoietic Prostaglandin D2 Synthase in Allergic Inflammation.
Cells 2019,
8, 619. doi:10.3390/cells8060619.
[Google Scholar]
126.
Chrysavgis LG, Kazanas S, Bafa K, Rozani S, Koloutsou M-E, Cholongitas E. Glucagon-like Peptide 1, Glucose-Dependent Insulinotropic Polypeptide, and Glucagon Receptor Agonists in Metabolic Dysfunction-Associated Steatotic Liver Disease: Novel Medication in New Liver Disease Nomenclature.
Int. J. Mol. Sci. 2024,
25, 3832. doi:10.3390/ijms25073832.
[Google Scholar]
127.
Coskun T, Sloop KW, Loghin C, Alsina-Fernandez J, Urva S, Bokvist KB, et al. LY3298176, a novel dual GIP and GLP-1 receptor agonist for the treatment of type 2 diabetes mellitus: From discovery to clinical proof of concept.
Mol. Metab. 2018,
18, 3–14. doi:10.1016/j.molmet.2018.09.009.
[Google Scholar]
128.
Zimmermann T, Thomas L, Baader-Pagler T, Haebel P, Simon E, Reindl W, et al. BI 456906: Discovery and preclinical pharmacology of a novel GCGR/GLP-1R dual agonist with robust anti-obesity efficacy.
Mol Metab 2022,
66, 101633. doi:10.1016/j.molmet.2022.101633.
[Google Scholar]
129.
Harrison S, Tomah S, Suschak J, Roberts S, Yang J, He L, et al. Pemvidutide, a glp-1/glucagon dual receptor agonist, significantly reduces liver fat, fibro-inflammation, and body weight in patients with non-alcoholic fatty liver disease: A 24-week multicenter, randomized, double-blind, placebo-controlled trial.
J. Hepatol. 2023,
78, S54–S55. doi:10.1016/S0168-8278(23)00517-2.
[Google Scholar]
130.
To D, Shin J, Karanth S, Lin Y-H, Sosnovtseva S, Bell AC. 72-LB: DD01, a Novel Once-Weekly Dual GLP-1/Glucagon Receptor Agonist, Improves Metabolic Health and Achieves Rapid, Clinically Significant Reductions in Hepatic Steatosis following Only Four Weeks of Treatment and without the Need for Significant Weight Loss in Overweight/Diabetic Subjects with NAFLD.
Diabetes 2023,
72, 72-LB. doi:10.2337/db23-72-LB.
[Google Scholar]
131.
Henderson SJ, Konkar A, Hornigold DC, Trevaskis JL, Jackson R, Fritsch Fredin M, et al. Robust anti-obesity and metabolic effects of a dual GLP-1/glucagon receptor peptide agonist in rodents and non-human primates.
Diabetes Obes. Metab. 2016,
18, 1176–1190. doi:10.1111/dom.12735.
[Google Scholar]
132.
Choi J, Kim JK, Lee SM, Kwon H, Lee J, Bae S, et al. 1830-P: Therapeutic Effect of a Novel Long-Acting GLP-1/GIP/Glucagon Triple Agonist (HM15211) in CDHFD-Induced NASH and Fibrosis Mice.
Diabetes 2020,
69, 1830-P. doi:10.2337/db20-1830-P.
[Google Scholar]
133.
Urva S, Coskun T, Loh MT, Du Y, Thomas MK, Gurbuz S, et al. LY3437943, a novel triple GIP, GLP-1, and glucagon receptor agonist in people with type 2 diabetes: A phase 1b, multicentre, double-blind, placebo-controlled, randomised, multiple-ascending dose trial.
Lancet 2022,
400, 1869–1881. doi:10.1016/S0140-6736(22)02033-5.
[Google Scholar]
134.
Bossart M, Wagner M, Elvert R, Evers A, Hübschle T, Kloeckener T, et al. Effects on weight loss and glycemic control with SAR441255, a potent unimolecular peptide GLP-1/GIP/GCG receptor triagonist.
Cell Metab. 2022,
34, 59–74.e10. doi:10.1016/j.cmet.2021.12.005.
[Google Scholar]
135.
Loomba R, Hartman ML, Lawitz EJ, Vuppalanchi R, Boursier J, Bugianesi E, et al. Tirzepatide for Metabolic Dysfunction–Associated Steatohepatitis with Liver Fibrosis.
New Engl. J. Med. 2024,
391, 299–310. doi:10.1056/NEJMoa2401943.
[Google Scholar]
136.
Blüher M, Rosenstock J, Hoefler J, Manuel R, Hennige AM. Dose–response effects on HbA1c and bodyweight reduction of survodutide, a dual glucagon/GLP-1 receptor agonist, compared with placebo and open-label semaglutide in people with type 2 diabetes: A randomised clinical trial.
Diabetologia 2024,
67, 470–482. doi:10.1007/s00125-023-06053-9.
[Google Scholar]
137.
Wang DD, Naumova AV, Isquith D, Sapp J, Huynh KA, Tucker I, et al. Dapagliflozin reduces systemic inflammation in patients with type 2 diabetes without known heart failure.
Cardiovasc. Diabetol. 2024,
23, 197. doi:10.1186/s12933-024-02294-z.
[Google Scholar]
138.
Heerspink HJL, Stefánsson BV, Correa-Rotter R, Chertow GM, Greene T, Hou F-F, et al. Dapagliflozin in Patients with Chronic Kidney Disease.
New Engl. J. Med. 2020,
383, 1436–1446. doi:10.1056/NEJMoa2024816.
[Google Scholar]
139.
Perkovic V, Jardine MJ, Neal B, Bompoint S, Heerspink HJL, Charytan DM, et al. Canagliflozin and Renal Outcomes in Type 2 Diabetes and Nephropathy.
New Engl. J. Med. 2019,
380, 2295–2306. doi:10.1056/NEJMoa1811744.
[Google Scholar]
140.
Yan C, Boyd DD. Regulation of matrix metalloproteinase gene expression.
J. Cell Physiol. 2007,
211, 19–26. doi:10.1002/jcp.20948.
[Google Scholar]
141.
Fanjul-Fernández M, Folgueras AR, Cabrera S, López-Otín C. Matrix metalloproteinases: Evolution, gene regulation and functional analysis in mouse models.
Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 2010,
1803, 3–19. doi:10.1016/j.bbamcr.2009.07.004.
[Google Scholar]
142.
Samuel CS, Bennett RG. Relaxin as an anti-fibrotic treatment: Perspectives, challenges and future directions.
Biochem. Pharmacol. 2022,
197, 114884. doi:10.1016/j.bcp.2021.114884.
[Google Scholar]
143.
Lauer D, Slavic S, Sommerfeld M, Thöne-Reineke C, Sharkovska Y, Hallberg A, et al. Angiotensin Type 2 Receptor Stimulation Ameliorates Left Ventricular Fibrosis and Dysfunction via Regulation of Tissue Inhibitor of Matrix Metalloproteinase 1/Matrix Metalloproteinase 9 Axis and Transforming Growth Factor β1 in the Rat Heart.
Hypertension 2014,
63, e60–e67. doi:10.1161/HYPERTENSIONAHA.113.02522.
[Google Scholar]
144.
Koulis C, Chow BSM, McKelvey M, Steckelings UM, Unger T, Thallas-Bonke V, et al. AT2R Agonist, Compound 21, Is Reno-Protective Against Type 1 Diabetic Nephropathy.
Hypertension 2015,
65, 1073–1081. doi:10.1161/HYPERTENSIONAHA.115.05204.
[Google Scholar]
145.
Swaney J, Chapman C, Correa L, Stebbins K, Bundey R, Prodanovich P, et al. A novel, orally active LPA
1 receptor antagonist inhibits lung fibrosis in the mouse bleomycin model.
Br. J. Pharmacol. 2010,
160, 1699–1713. doi:10.1111/j.1476-5381.2010.00828.x.
[Google Scholar]
146.
Haga A, Nagai H, Deyashiki Y. Autotaxin Promotes the Expression of Matrix Metalloproteinase-3 via Activation of the MAPK Cascade in Human Fibrosarcoma HT-1080 Cells.
Cancer Investig. 2009,
27, 384–390. doi:10.1080/07357900802491469.
[Google Scholar]
147.
Jin Y, Han H-C, Lindsey ML. ACE inhibitors to block MMP-9 activity: New functions for old inhibitors.
J. Mol. Cell Cardiol. 2007,
43, 664–666. doi:10.1016/j.yjmcc.2007.09.002.
[Google Scholar]
148.
Loomba R, Mohseni R, Lucas KJ, Gutierrez JA, Perry RG, Trotter JF, et al. TVB-2640 (FASN Inhibitor) for the Treatment of Nonalcoholic Steatohepatitis: FASCINATE-1, a Randomized, Placebo-Controlled Phase 2a Trial.
Gastroenterology 2021,
161, 1475–1486. doi:10.1053/j.gastro.2021.07.025.
[Google Scholar]
149.
Khalil N, Manganas H, Ryerson CJ, Shapera S, Cantin AM, Hernandez P, et al. Phase 2 clinical trial of PBI-4050 in patients with idiopathic pulmonary fibrosis.
Eur. Respir. J. 2019,
53, 1800663. doi:10.1183/13993003.00663-2018.
[Google Scholar]
150.
Harrison SA, Mayo PR, Hobbs TM, Canizares C, Foster EP, Zhao C, et al. Rencofilstat, a cyclophilin inhibitor: A phase 2a, multicenter, single-blind, placebo-controlled study in F2/F3 NASH.
Hepatol. Commun. 2022,
6, 3379–3392. doi:10.1002/hep4.2100.
[Google Scholar]
151.
Khanna D, Clements PJ, Furst DE, Korn JH, Ellman M, Rothfield N, et al. Recombinant human relaxin in the treatment of systemic sclerosis with diffuse cutaneous involvement: A randomized, double-blind, placebo-controlled trial.
Arthritis Rheum. 2009,
60, 1102–1111. doi:10.1002/art.24380.
[Google Scholar]
152.
Chalasani N, Abdelmalek MF, Garcia-Tsao G, Vuppalanchi R, Alkhouri N, Rinella M, et al. Effects of Belapectin, an Inhibitor of Galectin-3, in Patients With Nonalcoholic Steatohepatitis With Cirrhosis and Portal Hypertension.
Gastroenterology 2020,
158, 1334–1345.e5. doi:10.1053/j.gastro.2019.11.296.
[Google Scholar]
153.
Climax J, Newsome PN, Hamza M, Weissbach M, Coughlan D, Sattar N, et al. Effects of Epeleuton, a Novel Synthetic Second-Generation n-3 Fatty Acid, on Non-Alcoholic Fatty Liver Disease, Triglycerides, Glycemic Control, and Cardiometabolic and Inflammatory Markers.
J. Am. Heart Assoc. 2020,
9, e016334. doi:10.1161/JAHA.119.016334.
[Google Scholar]
154.
Calle RA, Amin NB, Carvajal-Gonzalez S, Ross TT, Bergman A, Aggarwal S, et al. ACC inhibitor alone or co-administered with a DGAT2 inhibitor in patients with non-alcoholic fatty liver disease: Two parallel, placebo-controlled, randomized phase 2a trials.
Nat. Med. 2021,
27, 1836–1848. doi:10.1038/s41591-021-01489-1.
[Google Scholar]
155.
Safadi R, Braun M, Francis A, Milgrom Y, Massarwa M, Hakimian D, et al. Randomised clinical trial: A phase 2 double-blind study of namodenoson in non-alcoholic fatty liver disease and steatohepatitis.
Aliment. Pharmacol. Ther. 2021,
54, 1405–1415. doi:10.1111/apt.16664.
[Google Scholar]
156.
Strambu IR, Seemayer CA, Fagard LM-CA, Ford PA, Van der Aa TAK, de Haas-Amatsaleh AA, et al. GLPG1205 for idiopathic pulmonary fibrosis: A phase 2 randomised placebo-controlled trial.
Eur. Respir. J. 2023,
61, 2201794. doi:10.1183/13993003.01794-2022.
[Google Scholar]
157.
Maher TM, Ford P, Brown KK, Costabel U, Cottin V, Danoff SK, et al. Ziritaxestat, a Novel Autotaxin Inhibitor, and Lung Function in Idiopathic Pulmonary Fibrosis.
JAMA 2023,
329, 1567. doi:10.1001/jama.2023.5355.
[Google Scholar]
158.
Maron MS, Chan RH, Kapur NK, Jaffe IZ, McGraw AP, Kerur R, et al. Effect of Spironolactone on Myocardial Fibrosis and Other Clinical Variables in Patients with Hypertrophic Cardiomyopathy.
Am. J. Med. 2018,
131, 837–841. doi:10.1016/j.amjmed.2018.02.025.
[Google Scholar]
159.
Bonella F, Spagnolo P, Ryerson C. Current and Future Treatment Landscape for Idiopathic Pulmonary Fibrosis.
Drugs 2023,
83, 1581–1593. doi:10.1007/s40265-023-01950-0.
[Google Scholar]
160.
Brennan PN, Elsharkawy AM, Kendall TJ, Loomba R, Mann DA, Fallowfield JA. Antifibrotic therapy in nonalcoholic steatohepatitis: Time for a human-centric approach.
Nat. Rev. Gastroenterol. Hepatol. 2023,
20, 679–688. doi:10.1038/s41575-023-00796-x.
[Google Scholar]
161.
Zhang C-Y, Liu S, Yang M. Treatment of liver fibrosis: Past, current, and future.
World J. Hepatol. 2023,
15, 755–774. doi:10.4254/wjh.v15.i6.755.
[Google Scholar]
162.
Ruiz-Ortega M, Lamas S, Ortiz A. Antifibrotic Agents for the Management of CKD: A Review.
Am. J. Kidney Dis. 2022,
80, 251–263. doi:10.1053/j.ajkd.2021.11.010.
[Google Scholar]
163.
Fang L, Murphy AJ, Dart AM. A Clinical Perspective of Anti-Fibrotic Therapies for Cardiovascular Disease.
Front. Pharmacol. 2017,
8, 186. doi:10.3389/fphar.2017.00186.
[Google Scholar]
164.
Lescoat A, Varga J, Matucci-Cerinic M, Khanna D. New promising drugs for the treatment of systemic sclerosis: Pathogenic considerations, enhanced classifications, and personalized medicine.
Expert. Opin. Investig. Drugs 2021,
30, 635–652. doi:10.1080/13543784.2021.1923693.
[Google Scholar]
165.
Li X, Zhu L, Wang B, Yuan M, Zhu R. Drugs and Targets in Fibrosis.
Front. Pharmacol. 2017,
8, 855. doi:10.3389/fphar.2017.00855.
[Google Scholar]
166.
Fuster-Martínez I, Calatayud S. The current landscape of antifibrotic therapy across different organs: A systematic approach.
Pharmacol. Res. 2024,
205, 107245. doi:10.1016/j.phrs.2024.107245.
[Google Scholar]
167.
Zhao M, Wang L, Wang M, Zhou S, Lu Y, Cui H, et al. Targeting fibrosis: Mechanisms and clinical trials.
Signal Transduct. Target. Ther. 2022,
7, 206. doi:10.1038/s41392-022-01070-3.
[Google Scholar]
168.
Fernando K, Menon S, Jansen K, Naik P, Nucci G, Roberts J, et al. Achieving end-to-end success in the clinic: Pfizer’s learnings on R&D productivity.
Drug Discov. Today 2022,
27, 697–704. doi:10.1016/j.drudis.2021.12.010.
[Google Scholar]
169.
Pammolli F, Righetto L, Abrignani S, Pani L, Pelicci PG, Rabosio E. The endless frontier? The recent increase of R&D productivity in pharmaceuticals.
J. Transl. Med. 2020,
18, 162. doi:10.1186/s12967-020-02313-z.
[Google Scholar]
170.
Waring MJ, Arrowsmith J, Leach AR, Leeson PD, Mandrell S, Owen RM, et al. An analysis of the attrition of drug candidates from four major pharmaceutical companies.
Nat. Rev. Drug Discov. 2015,
14, 475–486. doi:10.1038/nrd4609.
[Google Scholar]
171.
Travers JG, Tharp CA, Rubino M, McKinsey TA. Therapeutic targets for cardiac fibrosis: From old school to next-gen. J. Clin. Investig. 2022, 132. doi:10.1172/JCI148554.
172.
Drenth JPH, Schattenberg JM. The nonalcoholic steatohepatitis (NASH) drug development graveyard: Established hurdles and planning for future success.
Expert. Opin. Investig. Drugs 2020,
29, 1365–1375. doi:10.1080/13543784.2020.1839888.
[Google Scholar]
173.
Raghu G. Idiopathic pulmonary fibrosis: Lessons from clinical trials over the past 25 years.
Eur. Respir. J. 2017,
50, 1701209. doi:10.1183/13993003.01209-2017.
[Google Scholar]
174.
Ilg MM, Stafford SJ, Mateus M, Bustin SA, Carpenter MJ, Muneer A, et al. Phosphodiesterase Type 5 Inhibitors and Selective Estrogen Receptor Modulators Can Prevent But Not Reverse Myofibroblast Transformation in Peyronie’s Disease.
J. Sex. Med. 2020,
17, 1848–1864. doi:10.1016/j.jsxm.2020.06.022.
[Google Scholar]
175.
Lapthorn AR, Ilg MM, Dziewulski P, Cellek S. Hydroxypyridone anti-fungals selectively induce myofibroblast apoptosis in an in vitro model of hypertrophic scars.
Eur. J. Pharmacol. 2024,
967, 176369. doi:10.1016/j.ejphar.2024.176369.
[Google Scholar]
176.
Cantor J. Maximizing the Therapeutic Effect of Endothelin Receptor Antagonists in Pulmonary Fibrosis: A Paradigm for Treating the Disease.
Int. J. Mol. Sci. 2024,
25, 4184. doi:10.3390/ijms25084184.
[Google Scholar]
177.
Shirakami E, Yamakawa S, Hayashida K. Strategies to prevent hypertrophic scar formation: A review of therapeutic interventions based on molecular evidence.
Burns Trauma. 2020,
8, tkz003. doi:10.1093/burnst/tkz003.
[Google Scholar]
178.
Alsomali H, Palmer E, Aujayeb A, Funston W. Early Diagnosis and Treatment of Idiopathic Pulmonary Fibrosis: A Narrative Review.
Pulm. Ther. 2023,
9, 177–193. doi:10.1007/s41030-023-00216-0.
[Google Scholar]
179.
Ginès P, Castera L, Lammert F, Graupera I, Serra-Burriel M, Allen AM, et al. Population screening for liver fibrosis: Toward early diagnosis and intervention for chronic liver diseases.
Hepatology 2022,
75, 219–228. doi:10.1002/hep.32163.
[Google Scholar]
180.
Sweeney M, Corden B, Cook SA. Targeting cardiac fibrosis in heart failure with preserved ejection fraction: Mirage or miracle?
EMBO Mol. Med. 2020,
12, e10865. doi:10.15252/emmm.201910865.
[Google Scholar]
181.
Ralph DJ, Brooks MD, Bottazzo GF, Pryor JP. The treatment of Peyronie’s disease with tamoxifen.
Br. J. Urol. 1992,
70, 648–651. doi:10.1111/j.1464-410X.1992.tb15836.x.
[Google Scholar]
182.
Megson M, Merrett C, Ilg MM, Cellek S, Ralph D. Can tamoxifen and a PDE5 inhibitor slow the progression of Peyronie’s disease?
J. Sex. Med. 2022,
19, S15–S16. doi:10.1016/j.jsxm.2022.08.158.
[Google Scholar]
183.
Geelhoed JP, de Jong IJ, Beck JJ. Treatment of acute phase of Peyronie’s disease with tamoxifen and tadalafil off label. In Proceedings of the Annual Congress of European Society for Sexual Medicine, Rotterdam, The Netherlands, 16–18 February 2023.
184.
Cellek S, Megson M, Ilg MM, Ralph DJ. A combination of phosphodiesterase type 5 inhibitor and tamoxifen for acute Peyronie’s disease: The first clinical signals.
J. Sex. Med. 2023,
20, 1057–1059. doi:10.1093/jsxmed/qdad083.
[Google Scholar]
185.
Teloken C, Rhoden EL, Grazziotin TM, Da Ros CT, Sogari PR, Souto CAV. Tamoxifen versus placebo in the treatment of Peyronie’s disease.
J. Urol. 1999,
162, 2003–2005. doi:10.1016/S0022-5347(05)68087-1.
[Google Scholar]
186.
Sanyal AJ, Brunt EM, Kleiner DE, Kowdley KV, Chalasani N, Lavine JE, et al. Endpoints and clinical trial design for nonalcoholic steatohepatitis.
Hepatology 2011,
54, 344–353. doi:10.1002/hep.24376.
[Google Scholar]
187.
Ding Y, Wang Y, Zhang W, Jia Q, Wang X, Li Y, et al. Roles of Biomarkers in Myocardial Fibrosis.
Aging Dis. 2020,
11, 1157. doi:10.14336/AD.2020.0604.
[Google Scholar]
188.
Wilkinson HN, Hardman MJ. Wound healing: Cellular mechanisms and pathological outcomes.
Open Biol. 2020,
10, 200223. doi:10.1098/rsob.200223.
[Google Scholar]
189.
Martinez Besteiro E, Beceiro MJ, Churruca Arróspide M, Mouhtar D, Peláez A, Alonso T, et al. Combination antifibrotic therapy in Idiopathic Pulmonary Fibrosis patients: Real life experience in an ILD Unit. In 12.01—Idiopathic Interstitial Pneumonias; European Respiratory Society: Lausanne, Switzerland, 2022; p. 3886. doi:10.1183/13993003.congress-2022.3886.
190.
Swinney DC, Anthony J. How were new medicines discovered?
Nat. Rev. Drug Discov. 2011,
10, 507–519. doi:10.1038/nrd3480.
[Google Scholar]
191.
Lapthorn AR, Ilg MM, Sullivan JV, Dziewulski P, Cellek S. Phenotypic screening identifies hydroxypyridone anti-fungals as novel medicines for the prevention of hypertrophic scars.
Eur. J. Pharmacol. 2022,
937, 175374. doi:10.1016/j.ejphar.2022.175374.
[Google Scholar]
192.
Gerckens M, Schorpp K, Pelizza F, Wögrath M, Reichau K, Ma H, et al. Phenotypic drug screening in a human fibrosis model identified a novel class of antifibrotic therapeutics.
Sci. Adv. 2021,
7, eabb3673. doi:10.1126/sciadv.abb3673.
[Google Scholar]
193.
Yu S, Chen E, Sherwood L, Hull M, Woods AK, Tremblay MS. Ex. Vivo Cell-Based Screening Platform for Modulators of Hepatosteatosis.
ACS Chem. Biol. 2017,
12, 1937–1946. doi:10.1021/acschembio.7b00420.
[Google Scholar]
194.
Garcia H, Serafin AS, Silbermann F, Porée E, Viau A, Mahaut C, et al. Agonists of prostaglandin E
2 receptors as potential first in class treatment for nephronophthisis and related ciliopathies.
Proc. Natl. Acad. Sci. USA 2022,
119, e2115960119. doi:10.1073/pnas.2115960119.
[Google Scholar]
195.
Reid BG, Stratton MS, Bowers S, Cavasin MA, Demos-Davies KM, Susano I, et al. Discovery of novel small molecule inhibitors of cardiac hypertrophy using high throughput, high content imaging.
J. Mol. Cell Cardiol. 2016,
97, 106–113. doi:10.1016/j.yjmcc.2016.04.015.
[Google Scholar]
196.
Ilg MM, Lapthorn AR, Ralph DJ, Cellek S. Phenotypic screening of 1,953 FDA-approved drugs reveals 26 hits with potential for repurposing for Peyronie’s disease.
PLoS ONE 2022,
17, e0277646. doi:10.1371/journal.pone.0277646.
[Google Scholar]