
Part of Special Issue:
Synthetic Biology in the Manufacturing of Chemicals and Fuels
Download PDF
Cite This Article
Navigable Genome Engineering: Stepwise Correlation for Precision-Guided Optimization of Microbial Cell Factory Phenotypes

Author Information
Other Information
1
MOE Key Laboratory for Industrial Biocatalysis, Institute of Biochemical Engineering, Department of Chemical Engineering, Tsinghua University, Beijing 100084, China
2
Center for Synthetic and Systems Biology, Tsinghua University, Beijing 100084, China
*
Authors to whom correspondence should be addressed.
Received: 20 December 2024 Accepted: 10 March 2025 Published: 17 March 2025

© 2025 The authors. This is an open access article under the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/).
Views:552
Downloads:111
Synth. Biol. Eng.
2025,
3(1), 10003;
DOI: 10.70322/sbe.2025.10003
ABSTRACT:
Microbial cell factories, akin to “chips” in
biomanufacturing, concentrate the most intricate scientific challenges,
technical bottlenecks, and densest intellectual property. However, despite
extensive efforts in rational engineering, the inherent complexity of biological
systems and the limited knowledge of their underlying mechanisms still incur
substantial trial-and-error costs. This Perspective seeks to explore the
potential of a prior-knowledge-independent approach for optimizing microbial
cell factory phenotypes. We discuss the feasibility of stepwise genotypic
navigation in genome engineering and emphasize its ability to generate
high-quality genotype–phenotype association
data, thereby advancing AI-assisted genome modeling and further enabling
precision-guided optimization.
Keywords:
Microbial
cell factories; Phenotypic optimization; Genome engineering; Stepwise genotypic navigation
Synthetic biology has revolutionized industrial biotechnology by enabling the rational design of microbial cell factories for specific applications. However, optimizing complex industrially relevant phenotypes—such as enhanced substrate utilization, productivity, and robustness—remains a significant challenge due to the limited understanding of cellular regulatory and metabolic networks. This knowledge gap constrains our ability to modulate these systems to achieve desired outcomes precisely.
1. Advances and Limitations of Global Mutagenesis in Phenotypic Optimization
Recent developments in in vivo global mutagenesis technologies have significantly enhanced the capacity for genome-wide diversification, driving the rapid evolution of microbial strains with improved phenotypes [1,2]. However, such global strategies are inherently limited in their ability to resolve genotype–phenotype relationships. Despite the integration of advanced multi-omics technologies [3], the prevalence of non-contributory mutations significantly complicates the process, rendering the functional validation of candidate mutations labor-intensive and reliant on extensive trial-and-error experimentation to determine their relevance to target traits. Consequently, global mutagenesis approaches are highly inefficient in exploring the vast genome sequence space, generating sparse datasets with minimal actionable insights. These limitations severely constrain their ability to guide the further optimization of microbial cell factory phenotypes, underscoring the need for more targeted and precise methods to navigate genotype–phenotype landscapes.
2. Inspiration from Protein Evolution: Sequence Space Compression
The concept of sequence space compression [4] in protein engineering provides a valuable framework for addressing the complexity of genome-scale evolutionary engineering. Protein evolution is inherently constrained by the vast, high-dimensional fitness landscape, which remains computationally and experimentally intractable for exhaustive exploration [5]. For instance, a small protein with 100 amino acids presents an astronomical sequence space of 20100 (~10130) at the amino acid level, which expands to 4300 (~10181) at the nucleotide level. Current directed evolution methods are capable of identifying isolated high-fitness sequences; however, they probe only a minute fraction of this complex landscape, leaving its structural principles and functional relationships largely unexplored. To address this challenge, an evolutionary scanning method, EvoScan, has recently been developed by replacing the global mutagenesis tool MP6 in phage-assisted evolution with the targeted and segmented mutagenesis system, EvolvR. This innovation allows for systematic segmentation and exhaustive scanning of the protein sequence space, enabling the identification of high-fitness sequence anchors. By integrating deep learning and large language models, EvoScan further facilitates the exploration of genotype–phenotype relationships within high-dimensional mutational combinations. These advancements significantly enhance the capacity for rational protein function design, bridging the gap between directed evolution and computational predictive models.
Applying sequence space compression to the genome extends the principles of protein evolution to address an even larger and more complex sequence landscape. For instance, a bacterial genome of 3000 genes theoretically presents a combinatorial space of ~101431 if each gene is modeled in three functional states (unchanged, upregulated, or downregulated). Most genetic changes, however, have negligible effects on target phenotypes. Thus, by identifying key functional genes strongly associated with phenotypes of interest (POIs)—referred to as gene-level anchors—and excluding genes with lower associations, the dimensionality of the genotype space can be significantly reduced (Figure 1). This facilitates computational prediction of optimal combinatorial modifications, which can subsequently be refined through in situ mutagenesis to identify base-level anchors at single-nucleotide resolution.
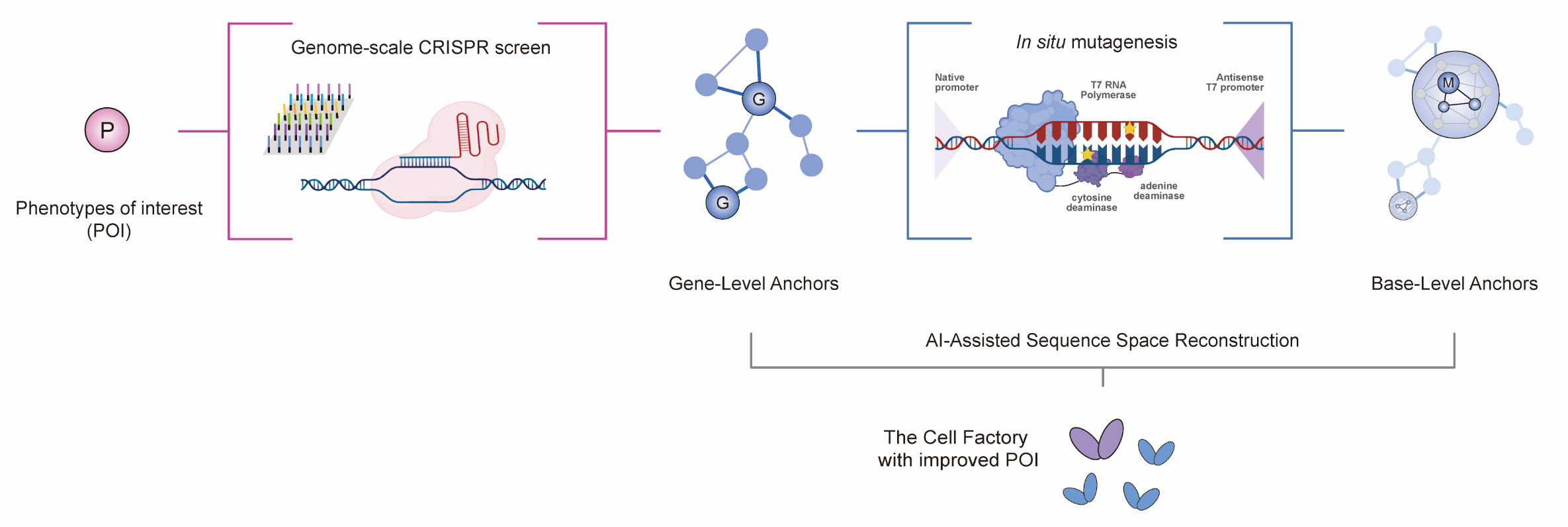
Figure 1. Schematic representation of navigable genome engineering. This concept involves identifying gene-level anchors through functional genomics screens, exemplified in the figure by genome-scale CRISPR screening, and base-level anchors through <i>in situ</i> mutagenesis, illustrated with the MutaT7 technology. These anchors provide highly relevant mutation data for AI-assisted genome modeling, enabling precision-guided optimization of microbial cell factory phenotypes.
3. Navigating Gene-Level Anchors
Mapping POIs from whole-genome sequences to gene-level anchors aligns with the core objective of functional genomics screens [6]—identifying genes that contribute to a phenotype. However, to effectively compress and reconstruct the genome sequence space for POI optimization, it is imperative to go beyond individual gene associations and emphasize gene-gene interactions (epistasis). The key challenge is understanding how simultaneous perturbations across multiple genes influence a phenotype, a critical requirement for capturing the functional complexity of the genome.
Traditional approaches such as knockout collections, overexpression libraries, RNA interference (RNAi), and transposon sequencing (Tn-seq) have provided significant insights into gene function. However, these methods are not inherently designed for systematic multi-gene combination analysis.
-
- RNAi: Often suffers from off-target effects, reducing precision.
-
- Overexpression libraries: Limited by vector capacity, hindering scalability.
-
- Knockout/Tn-seq: Primarily focus on single-gene disruptions, failing to capture combinatorial effects.
-
- Gene KO screens (CRISPR-Cas)
-
- Gene activation and interference screens (CRISPRa/i)
-
- Prematurely terminated ORF screens (Base editor) [9]
-
- Gene overexpression screens (CRISPR-associated transposons) [10]
4. Navigating Base-Level Anchors
Identifying base-level anchors involves in situ mutagenesis to refine gene-level findings and uncover critical nucleotide changes influencing POIs (Figure 2). This approach resembles early multiplex automated genome engineering (MAGE) [14], which introduced targeted mutations into pathway-specific genes to optimize traits such as lycopene biosynthesis. However, unlike traditional methods focused solely on genes within known pathways, the strategy proposed here identifies mutational targets from genome-wide gene-level anchors associated with POIs.
Despite its utility, MAGE suffers from low ssDNA incorporation efficiency and operational challenges associated with continuous mutagenesis, leading to sparse datasets for base-level mutations.
Recent advancements have introduced alternative systems to overcome these limitations:
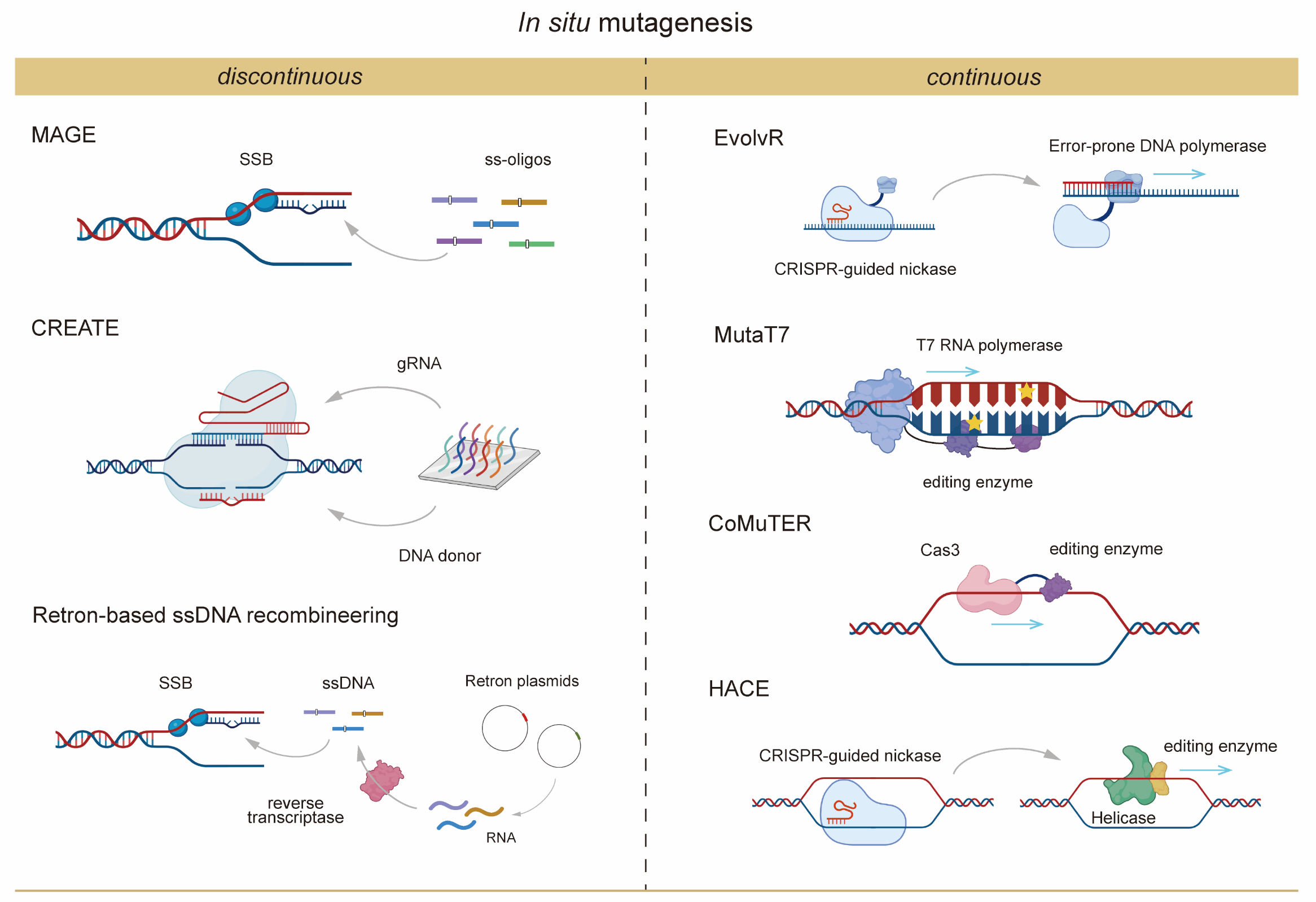
Figure 2. Schematic diagram of representative discontinuous and continuous <i>in situ</i> mutagenesis techniques. Abbreviations include: SSB (single-stranded binding protein), ss-oligos (single-stranded oligos), ssDNA (single-stranded DNA).
-
- CREATE [15] (CRISPR-enabled trackable genome engineering): CREATE integrates CRISPR-directed DNA cleavage with mutation-bearing donor recombination, offering a more efficient and trackable approach to genome engineering.
-
- Retron-based ssDNA recombineering [16]: Retron systems facilitate the endogenous synthesis of ssDNA, addressing the low efficiency of traditional MAGE. This approach enables the simultaneous editing of up to four loci in E. coli, providing a scalable and efficient platform for multi-site genome editing [17].
-
- EvolvR: Utilizes a fusion of nicking Cas9 (nCas9) with an error-prone DNA polymerase to introduce mutations within a defined window of approximately 56 nucleotides near the targeted site.
-
- MutaT7: Employs a fusion of T7 RNA polymerase with a deaminase to induce mutations in regions downstream of T7 promoters.
-
- CoMuTER: Leverages a fusion of Cas3 helicase/nuclease with a deaminase to unwind DNA and introduce mutations over extensive regions, up to 55 kilobases.
-
- HACE: Utilizes nCas9 to recruit a helicase-deaminase fusion enzyme, enabling locus-specific mutagenesis across regions exceeding 1000 base pairs.
5. AI-Assisted Sequence Space Reconstruction
Machine learning models have been widely applied in metabolic engineering to optimize pathway configurations, linking combinatorial modifications (e.g., promoter and ribosome binding site tuning) to production outcomes [23]. Applying this principle to genome-wide optimization, AI-assisted reconstruction can predict the combinatorial effects of gene-level anchors on POIs. Furthermore, multiplexed continuous evolution of top-scoring gene combinations has the potential to generate high-dimensional base-level mutational datasets, which can be leveraged to reconstruct the sequence space and guide navigation toward improved POIs. Compared to datasets generated through random mutagenesis, the high-correlation datasets derived from stepwise navigation of gene- and base-level anchors provide a more robust and structured foundation for predictive modeling and phenotype optimization.
It is critical to distinguish that protein sequence space compression operates at the amino acid level, linking “mutation combinations” to “functional changes” within a single protein. In contrast, POI-driven genome sequence space compression functions at the DNA level, correlating “mutation combinations” across multiple gene-level anchors to “changes in POIs”. AI models have already demonstrated success in capturing complex patterns in viral and bacterial genomes with single-nucleotide resolution [24,25], underscoring their potential to navigate and optimize high-dimensional genomic landscapes.
6. Conclusions and Outlook
The proposed concept of stepwise genotypic navigation integrates sequence space compression via multiplexed genome perturbation and mutagenesis with AI-assisted modeling. By navigating complex cell factory phenotypes to key functional genes and mutations, and exploring their interactions with the aid of AI, this approach holds promise for providing rapid guidance in the construction and optimization of cell factories. While current limitations in genome-wide multiplexing and high-dimensional data acquisition pose significant challenges, existing technologies offer a pathway for incremental progress. By leveraging sparse but high-correlation datasets, this approach establishes a foundation for the rational engineering of microbial strains tailored for industrial applications. Future advances in continuous evolution tools and machine learning will further enhance the precision and scalability of this strategy, transforming the landscape of cell factory engineering.
Acknowledgments
We thank members of the laboratory for helpful discussions.
Author Contributions
Conceptualization, C.Z.; Writing—Original Draft Preparation, X.Y.; Writing—Review & Editing, J.G. and J.S.; Project Administration, C.Z.; Funding Acquisition, C.Z.
Ethics Statement
Informed Consent Statement
Not applicable.
Data Availability Statement
Funding
This research received funding from the National Key Research and Development Program of China (2023YFC3402300), National Natural Science Foundation of China (U2032210), R&D projects in key areas of Guangdong Province (2022B1111050002) and the R&D projects of Hebei Province (22375503D).
Declaration of Competing Interest
The authors declare no competing financial interest.
References
1.
Wu YN, Jameel A, Xing XH, Zhang C. Advanced strategies and tools to facilitate and streamline microbial adaptive laboratory evolution. Trends Biotechnol. 2022, 40, 38–59. [Google Scholar]
2.
Simon AJ, d’Oelsnitz S, Ellington AD. Synthetic evolution. Nat. Biotechnol. 2019, 37, 730–743. [Google Scholar]
3.
Yang YF, Qiu MY, Yang Q, Wang Y, Wei H, Yang SH. Connecting Microbial Genotype with Phenotype in the Omics Era. Metab. Pathw. Eng. 2020, 2096, 217–233. [Google Scholar]
4.
Ma ZY, Li WJ, Shen YH, Xu YX, Liu GJ, Chang JM, et al. EvoAI enables extreme compression and reconstruction of the protein sequence space. Nat. Methods 2024, 22, 102–112. [Google Scholar]
5.
Papkou A, Garcia-Pastor L, Escudero JA, Wagner A. A rugged yet easily navigable fitness landscape. Science 2023, 382, 6673. [Google Scholar]
6.
Przybyla L, Gilbert LA. A new era in functional genomics screens. Nat. Rev. Genet. 2022, 23, 89–103. [Google Scholar]
7.
Teng YX, Jiang T, Yan YJ. The expanded CRISPR toolbox for constructing microbial cell factories. Trends Biotechnol. 2024, 42, 104–118. [Google Scholar]
8.
Shalem O, Sanjana NE, Zhang F. High-throughput functional genomics using CRISPR-Cas9. Nat. Rev. Genet. 2015, 16, 299–311. [Google Scholar]
9.
Liu Y, Wang RY, Liu JH, Lu H, Li HR, Wang Y, et al. Base editor enables rational genome-scale functional screening for enhanced industrial phenotypes in Corynebacterium glutamicum. Sci. Adv. 2022, 8, 35. [Google Scholar]
10.
Banta AB, Myers KS, Ward RD, Cuellar RA, Place M, Freeh CC, et al. A Targeted Genome-scale Overexpression Platform for Proteobacteria. bioRxiv 2024. doi: 10.1101/2024.03.01.582922.
11.
Sun L, Zheng P, Sun J, Wendisch VF, Wang Y. Genome-scale CRISPRi screening: A powerful tool in engineering microbiology. Eng. Microbiol. 2023, 3, 100089–100089. [Google Scholar]
12.
Wu YK, Li Y, Jin K, Zhang LP, Li JH, Liu YF, et al. CRISPR-dCas12a-mediated genetic circuit cascades for multiplexed pathway optimization. Nat. Chem. Biol. 2023, 19, 367. [Google Scholar]
13.
Yin JA, Frick L, Scheidmann MC, Liu TT, Trevisan C, Dhingra A, et al. Arrayed CRISPR libraries for the genome-wide activation, deletion and silencing of human protein-coding genes. Nat. Biomed. Eng. 2025, 9, 127–148. [Google Scholar]
14.
Wang HH, Isaacs FJ, Carr PA, Sun ZZ, Xu G, Forest CR, et al. Programming cells by multiplex genome engineering and accelerated evolution. Nature 2009, 460, 894. [Google Scholar]
15.
Garst AD, Bassalo MC, Pines G, Lynch SA, Halweg-Edwards AL, Liu RM, et al. Genome-wide mapping of mutations at single-nucleotide resolution for protein, metabolic and genome engineering. Nat. Biotechnol. 2017, 35, 48–55. [Google Scholar]
16.
Liu W, Pan Y, Zhang Y, Dong C, Huang L, Lian J. Intracellularly synthesized ssDNA for continuous genome engineering. Trends Biotech. 2024, 7799, 293. doi:10.1016/j.tibtech.2024.10.011. [Google Scholar]
17.
Liu WQ, Zuo SQ, Shao YR, Bi K, Zhao JR, Huang L, et al. Retron-mediated multiplex genome editing and continuous evolution in Escherichia coli. Nucleic Acids Res. 2023, 51, 8293–8307. [Google Scholar]
18.
Xu P, Liu ZH, Liu Y, Ma HZ, Xu YY, Bao Y, et al. Genome-wide interrogation of gene functions through base editor screens empowered by barcoded sgRNAs. Nat. Biotechnol. 2021, 39, 1403. [Google Scholar]
19.
Halperin SO, Tou CJ, Wong EB, Modavi C, Schaffer DV, Dueber JE. CRISPR-guided DNA polymerases enable diversification of all nucleotides in a tunable window. Nature 2018, 560, 248. [Google Scholar]
20.
Moore CL, Papa LJ, Shoulders MD. A Processive Protein Chimera Introduces Mutations across Defined DNA Regions. J. Am. Chem. Soc. 2018, 140, 11560–11564. [Google Scholar]
21.
Zimmermann A, Prieto-Vivas JE, Cautereels C, Gorkovskiy A, Steensels J, Van de Peer Y, et al. A Cas3-base editing tool for targetable in vivo mutagenesis. Nat. Commun. 2023, 14, 3389. [Google Scholar]
22.
Chen XD, Chen Z, Wythes G, Zhang Y, Orr BC, Sun G, et al. Helicase-assisted continuous editing for programmable mutagenesis of endogenous genomes. Science 2024, 386, 6718. [Google Scholar]
23.
Deng HX, Yu H, Deng YW, Qiu YL, Li FF, Wang XR, et al. Pathway Evolution Through a Bottlenecking-Debottlenecking Strategy and Machine Learning-Aided Flux Balancing. Adv. Sci. 2024, 11, e2306935. [Google Scholar]
24.
Nguyen E, Poli M, Durrant MG, Kang B, Katrekar D, Li DB, et al. Sequence modeling and design from molecular to genome scale with Evo. Science 2024, 386, eado9336. [Google Scholar]
25.
Shao B, Yan J. A long-context language model for deciphering and generating bacteriophage genomes. Nat. Commun. 2024, 15, 9392–9392. [Google Scholar]