1. Introduction
Among biofuels, biobutanol stands out due to its superior fuel characteristics: it contains nearly 30% more energy than ethanol and offers combustion performance comparable to gasoline. Unlike bioethanol and biodiesel, biobutanol can be directly blended with gasoline or diesel, requiring minimal or no engine modifications [
1,
2]. A well-known method for producing n-butanol biotechnologically is the Acetone-Butanol-Ethanol (ABE) fermentation process [
3,
4]. Despite its long-standing recognition, the ABE fermentation process still faces several challenges that hinder its industrial feasibility [
5]. It suffers from expensive feedstock, low butanol yield, low productivity, and high operational and equipment costs, with added significant water consumption. Many efforts have been made to make the biobutanol production process more feasible [
6]. These range from process engineering to microbe strain engineering [
7,
8,
9]. This review provides an overview of recent advances and challenges in metabolic and protein engineering approaches made to improve the biobutanol production process focusing on the metabolically engineered
Clostridium acetobutylicum [
10],
Clostridium tyrobutyricum [
11], and
Escherichia coli [
12].
2. Metabolic Engineering for Biobutanol Production
n-Butanol is naturally produced in some solventogenic clostridia [
5]. There have also been extensive efforts to engineer various microorganisms for biobutanol production [
8,
9,
10]. We highlight recent works on engineering
C. acetobutylicum,
C. tyrobutyricum, and
E. coli for n-butanol production in , with detailed discussion given in this section.
.
Notable examples of n-butanol production by engineered strains of C. acetobutylicum, C. tyrobutyricum, and E. coli.
| Strain |
Engineering Strategy |
Fermentation Conditions |
Titer (g/L) |
Yield (g/g) |
Productivity (g/L·h) |
Reference |
| C. acetobutylicum JB200 |
ATCC 55025 mutant obtained from adaptive evolution |
Batch fermentation of glucose |
20.3 |
0.23 |
0.33 |
[13] |
| C. acetobutylicum HKKO |
ATCC 55025 with cac3319 KO to increase butanol tolerance |
Batch fermentation of glucose |
18.2 |
0.20 |
0.38 |
[14] |
| C. acetobutylicum HKKO-Δadc-adhE2 |
KO: adc; OE: adhE2 |
Batch fermentation of glucose |
19.7 |
0.26 |
0.41 |
[15] |
| C. acetobutylicum ΔhydA |
ATCC 55025 with hydA KO to increase NADH |
Batch fermentation of glucose with methyl viologen |
13.8 |
0.28 |
0.31 |
[16] |
| C. acetobutylicum CAB1060 |
OE: atoB, hbd(Ck), crt, bcd, etfA, etfB, adhE2
KO: ptb, buk, ctfAB, ldhA, rexA, thlA, hbd
|
Fed-batch fermentation of glucose with in-situ butanol separation by vacuum distillation |
550 |
0.35 |
14 |
[17] |
| C. tyrobutyricum Ack-adhE2 |
OE: adhE2 in Ack strain |
Batch fermentation of glucose |
10 |
0.27 |
0.03 |
[18] |
|
Mannitol |
16 |
0.31 |
0.06 |
|
|
20.5 |
0.33 |
0.32 |
|
|
Glucose |
14.5 |
0.28 |
0.13 |
[19] |
|
Cassava bagasse |
13 |
0.34 |
0.26 |
[20] |
|
Corn fiber, cotton stalk, soybean hull, sugarcane bagasse |
15 |
0.3 |
0.3 |
[21] |
| Ack-adhE2-ctfAB |
OE: adhE2, ctfAB |
Glucose |
12 |
0.26 |
0.35 |
[22] |
| Ack-adhE2-scrABK |
OE: adhE2, scrA, scrB, scrK |
Sucrose |
14.8 |
0.21 |
0.15 |
[23] |
|
|
Sugarcane juice |
12.8 |
0.21 |
0.53 |
|
| Ack-adhE2-agluI |
OE: adhE2, agluI |
Maltose |
17.2 |
0.2 |
0.29 |
[24] |
|
|
Soluble starch |
16.2 |
0.17 |
0.19 |
|
| Ack-adhE2-xylTBA |
OE: adhE2, xylT, xylA, xylB |
Glucose/Xylose |
12 |
0.12 |
0.17 |
[25] |
|
|
Soybean hull |
15.7 |
0.24 |
0.29 |
|
| Δcat1::adhE2 |
KO: cat1; OE: adhE2 |
Glucose |
26.2 |
0.23 |
0.16 |
[26] |
|
|
Paper mill sludge |
16.5 |
0.26 |
0.17 |
[27] |
| Ct-pMA12G |
OE: adhE2, GroESL |
Brown algae biomass hydrolysate |
12.2 |
0.34 |
0.15 |
[28] |
| E. coli JCL187 |
OE: atoB, hbd, crt, bcd, etfA, etfB, adhE2;
KO: adhE, ldhA, frdBC, pta, fnr
|
Semi-aerobic batch fermentation with rich media supplemented with glycerol. |
0.55 |
N/A |
N/A |
[29] |
| E. coli JCL299 |
OE: atoB, hbd, crt, ter, adhE2, fdh
KO: ldhA adhE frdBC pta
|
Fed-batch fermentation of glucose with continuous butanol removal |
15 |
0.28 |
0.18 |
[30] |
| E. coli EB243 |
OE: atoB, hbd, crt, ter, adhE2, fdh
KO: adhE, eutE, yqhD, ldhA, ack, pta, pykAF, mdh, frdABCD, yciA, hyc-hyp/fdhF
|
Batch fermentation of glucose in fermentor with aeration for the first 12 h |
20 |
0.34 |
0.32 |
[31] |
| E. coli MG1655 (pZS-tesBT, pETM-CSA-P) |
OE: tesBT, car, sfp, adhE2 |
Fed-batch fermentation of glucose |
2.8 |
0.10 |
0.047 |
[32] |
| E. coli RB02 ΔyqhD ΔeutE [yqeF+fucO+] |
OE: yqeF, fucO
KO: yqhD, eutE
|
Batch fermentation of glucose |
13.5 |
0.27 |
0.33 |
[33] |
2.1. Metabolic Pathway in C. acetobutylicum
Clostridium acetobutylicum is synonymous with industrial ABE fermentation. This microorganism is a strictly anaerobic, spore-forming bacterium known for its biphasic metabolism [
34]. This fermentation is characterized by two distinct phases: the acetogenesis phase (
i.e., acid production) and the solventogenesis phase (
i.e., solvent production). The initial phase is characterized by rapid cell growth, preceding the formation of carboxylic acids, predominantly acetic and butyric acid. Acid excretion decreases the external pH, and these acids induce the biosynthesis of solventogenic enzymes for the subsequent phase of fermentation [
35].
In solventogenesis, these acids are reassimilated and serve as co-substrates for solvent production [
36]. During the solventogenesis phase, acid production ceases, cell growth halts, and the pH of the medium slightly increases from acid uptake [
37]. Carbon and electron flow are redirected toward solvent formation once solventogenesis begins. The transition to solvent production is thought to be an adaptive response by cells to the low pH environment [
38]. n-Butanol is majorly produced in this fermentation, with acetone and ethanol as two minor byproducts, where acetone, n-butanol, and ethanol are produced in a 3:6:1 weight ratio.
C. acetobutylicum is a robust microorganism, versatile in its ability to utilize various carbon sources, and adaptable to carbohydrate availability and culture conditions [
39]. The acetogenesis and solventogenesis biochemical pathways have been well described, with several studies identifying factors influencing solvent production [
3].
C. acetobutylicum uses the Embden-Meyerhof-Parnas (EMP) pathway [
40] to break hexose sugars into pyruvate, and pentose sugars are metabolized through the pentose phosphate pathway, producing intermediates that enter glycolysis [
41]. Historically, n-butanol has been produced from starch and sugar-based feedstocks in industrial ABE fermentation by solventogenic clostridia, including
C. acetobutylicum, which is limited by low butanol titer (~12 g/L), yield (~0.2 g/g), and productivity (~0.25 g/L·h) [
5].
Redox balance plays a major role in
C. acetobutylicum metabolism and is a phenomenon that is continually being revealed. Recently, Foulquier et al., 2022 characterized the electron pathways used for n-butanol synthesis in
C. acetobutylicum that were previously unaccounted for [
42]. The central metabolism for
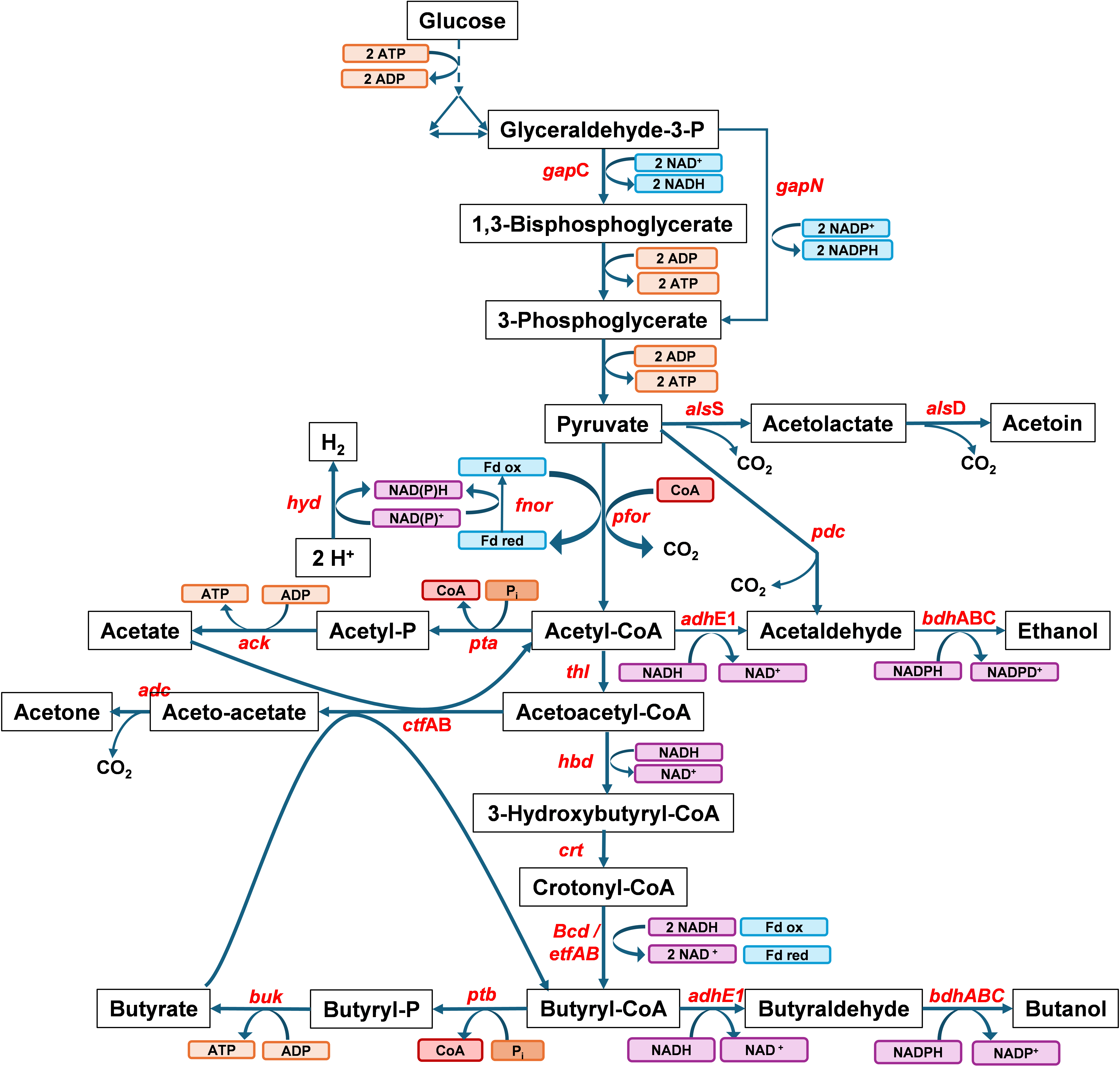
C. acetobutylicum is given in . From the EMP pathway, a single mole of glucose is converted to two moles of pyruvate and two moles of NADH, then two reduced ferredoxin and two acetyl-CoA molecules are produced during the decarboxylation of the two moles of pyruvate via pyruvate ferredoxin oxidoreductase (
pfor). However, during solventogenesis, the production of a single mole of n-butanol from the conversion of two moles of acetyl-CoA produces one mole of reduced ferredoxin generated when crotonyl-CoA is reduced to butyryl-CoA via butyryl-CoA dehydrogenase (BCD). Yet, four moles of NADH are consumed. Additionally, one mole of NADPH is consumed for butyraldehyde reduction via the NADPH-dependent alcohol dehydrogenases [
43]. Two moles of NADH are produced from the EMP pathway, and four moles of NADH are consumed for n-butanol production; therefore, the generation of two moles of NADH from the oxidation of two moles of reduced ferredoxin via ferredoxin-NAD
+ reductase is required. Additionally, the absence of an oxidative pentose-phosphate pathway [
44] and the low-level expression of
gapN, encoding the non-phosphorylating NADPH-producing glyceraldehyde-3-P dehydrogenase, means one mole of NADPH must be produced from the oxidation of reduced ferredoxin via ferredoxin-NADP
+ reductase. The study showed that ferredoxin-NADP
+-reductase inactivation considerably lowered butanol production and raised acetone production, whereas overexpression of this gene increased butanol production, resulting in 72% of the theoretical maximum butanol yield from glucose. These findings infer that NAD(P)H flux is a limiting factor to butanol production and demonstrate that electron flux manipulation modulates carbon fluxes.
. Central metabolism of <em>Clostridium acetobutylicum</em>. Genes in the pathways—<em>ack</em>: acetate kinase, <em>adc</em>: acetoacetate decarboxylase, <em>adh</em>E1: aldehyde dehydrogenase, <em>als</em>D: alpha-acetolactate decarboxylase, <em>als</em>S: acetolactate synthase, <em>bcd</em>: butyryl-CoA dehydrogenase, <em>bdh</em>: butanol dehydrogenase, <em>buk</em>: butyrate kinase, <em>crt</em>: crotonase, <em>ctf</em>AB: CoA-transferase, <em>etf</em>: electron transfer flavoprotein, <em>hbd</em>: 3-hydroxybutyryl-CoA dehydrogenase, <em>hyd</em>: hydrogenase,<em> fnor</em>: ferredoxin-NAD(P)<sup>+</sup> oxidoreductases, <em>pdc</em>: pyruvate decarboxylase, <em>pfor</em>: pyruvate:ferredoxin oxidoreductase, <em>pta</em>: phosphotransacetylase, <em>ptb</em>: phosphotransbutyrylase, <em>thl</em>: thiolase, <em>gap</em>C: NADH-dependent glyceraldehyde-3-phosphate dehydrogenase, <em>gap</em>N: nonphosphorylating NADPH-producing glyceraldehyde-3-phosphate dehydrogenase. Fd<sub>ox</sub> represents oxidized ferredoxin, Fd<sub>red</sub> represents reduced ferredoxin.
Many studies have investigated increasing n-butanol production through the metabolic engineering of
C. acetobutylicum. Some approaches include mutation and overexpression of thiolase (
thl) [
45], disruption of phosphotransacytylase (
pta) and butyrate kinase (
buk) [
46], and CoA transferase (
ctfAB) down-regulation combined with aldehyde/alcohol dehydrogenase (
adhE1,
adhE2) overexpression [
47]. However, perhaps the most comprehensive work done on the metabolic engineering of
C. acetobutylicum for enhancing n-butanol production was performed by Nguyen et al., 2018 [
17]. Nguyen and his team created a C.
acetobutylicum strain containing multiple gene deletions and heterologous gene substitutions. Gene knockouts include:
ptb (phosphotransbutyrylase) and
buk (butyrate kinase) encoding butyrate biosynthesis from butyryl-CoA;
ldhA, a gene encoding lactate production;
ctfAB, a gene encoding acetone formation; and
rexA, a redox-sensing transcriptional regulator recognized as gene repressor for the C4 formation pathway (
thlA,
crt-
bcd-
etfAB-
hbd, and
adhE2). Gene substitutions Δ
thlA::
atoB (acetyl-CoA acetyltransferase from
E. coli) and Δ
hbd::
hbd1 (NADPH-dependent 3-hydroxybutyryl-CoA dehydrogenase from
C. kluveris) were made to eliminate potential CoA-SH inhibition of the enzyme encoded by
thlA and to utilize the high NADPH/NADP
+ ratio driving force. This resulted in a strain which achieved 83% of the theoretical maximum butanol yield, and a sixfold butanol-to-ethanol ratio increase. However, attempts to eliminate genes
pta and
ack, responsible for acetate accumulation, were not successful.
Another pertinent study addressed a central issue described above: the limitation of reducing power in n-butanol production. The disruption of hydrogenase gene
hydA in
C. acetobutylicum increased intracellular reducing power and enhanced butanol production with significantly decreased by-products, achieving a high butanol yield of 0.28 g/g and butanol ratio of 84.0% when methyl viologen was added in the fermentation [
48]. In addition, researchers introduced a 2,3-butanediol synthesis pathway to mitigate the need for acetone production, which acted as an NADH-compensating module [
49]. This study illustrated the role of acetone production in regulating redox balance, where overexpression of a 2,3-butanediol production pathway nearly eliminated acetone production and resulted in a high total alcohol yield of 0.44 g/g glucose. Finally, butanol production was also enhanced through intracellular NADH regeneration in a CdSe-
C. acetobutylicumg semi-photosynthetic biohybrid system, which used light-activated cadmium selenide quantum dots (CdSe QDs) to capture electrons from light, resulting in a 45.5% increase in NADH/NAD
+ ratio compared to
C. acetobutylicum without CdSe QDs [
50]. The photo-fermentation system produced 14.8 g/L butanol at a 0.29 g/g yield from rice straw hydrolysate.
Butanol production has also been increased by overexpressing 6-phosphofructokinase (
pfkA) and pyruvate kinase (
pykA), two key genes in the glycolytic pathway, in ABE fermentation due to enhanced butanol tolerance [
51].
Strain degeneration due to loss of the pSOL1 megaplasmid containing the
sol operon genes responsible for solvent production in
C. acetobutylicum often limits its use in continuous ABE fermentation [
52]. This limitation can be mitigated by integrating the sol operon genes on the chromosome for stable gene expression for solvent production in a continuous fermentation process [
53].
In general, the ABE fermentation is unstable and difficult to control due to clostridia’s complex life cycle involving acidogenesis, solventogenesis, and sporulation, which are highly regulated by various kinases, transcription factors, and interlocking signal pathways [
54]. Under stress, the cells sporulate and halt their metabolism [
55]. Sporulation limits cell’s ability to produce butanol and can make clostridial fermentation challenging and prone to failure [
56]. Clostridia sporulation is regulated and controlled by Spo0A, which is phosphorylated/dephosphorylated by orphan histidine kinases encoded by
cac3319,
cac0903,
cac0323, and
cac0437 [
57,
58,
59]. A recent study showed that the knockout of
cac3319 aborted sporulation and enhanced butanol tolerance and production [
14]. In contrast, premature sporulation was observed with
cac0437 knockout, which also caused early autolysis, inhibited the transition from acidogenesis to solventogenesis, and resulted in poor butanol production [
60]. Finally, a mutant with double knockouts of
cac3319 and
cac0323 gave the highest butanol production of >20 g/L [
15]. These studies demonstrated that engineering the orphan histidine kinases can effectively regulate sporulation and increase butanol tolerance and production in
C. acetobutylicum. Further knockout of
adc and overexpression of
adhE2 in the mutant with histidine kinase genes (
cac3319 and
cac0323) knockout decreased acetone production to 0.1 g/L and increased butanol production to 19.7 g/L with a productivity of 0.41 g/L·h and yield of 0.26 g/g [
16].
Finally, engineering the accessory gene regulator (Agr) involved in the quorum sensing system, which regulates autoinducing peptides (AIPs) and plays a critical role in intercellular communication, may provide a novel strategy for optimizing cell metabolism and butanol biosynthesis in
Clostridium acetobutylicum [
61].
2.3. Metabolic Pathway in C. tyrobutyricum
Acidogenic clostridia contain metabolic pathways to produce acetic and butyric acids, which can be leveraged to produce biofuels, making them novel hosts for n-butanol production.
Clostridium tyrobutyricum is one such clostridia. Bao et al., 2020 have written a comprehensive review on n-butanol and butyrate production in
C. tyrobutyricum [
11]. It is a super butyrate-producer, which utilizes a butyryl-CoA/acetate CoA transferase (encoded by
cat1) to re-assimilate acetate for butyrate biosynthesis, unlike other clostridia, which typically use the phosphotransbutyrylase-butyrate kinase (
ptb-bk) pathway [
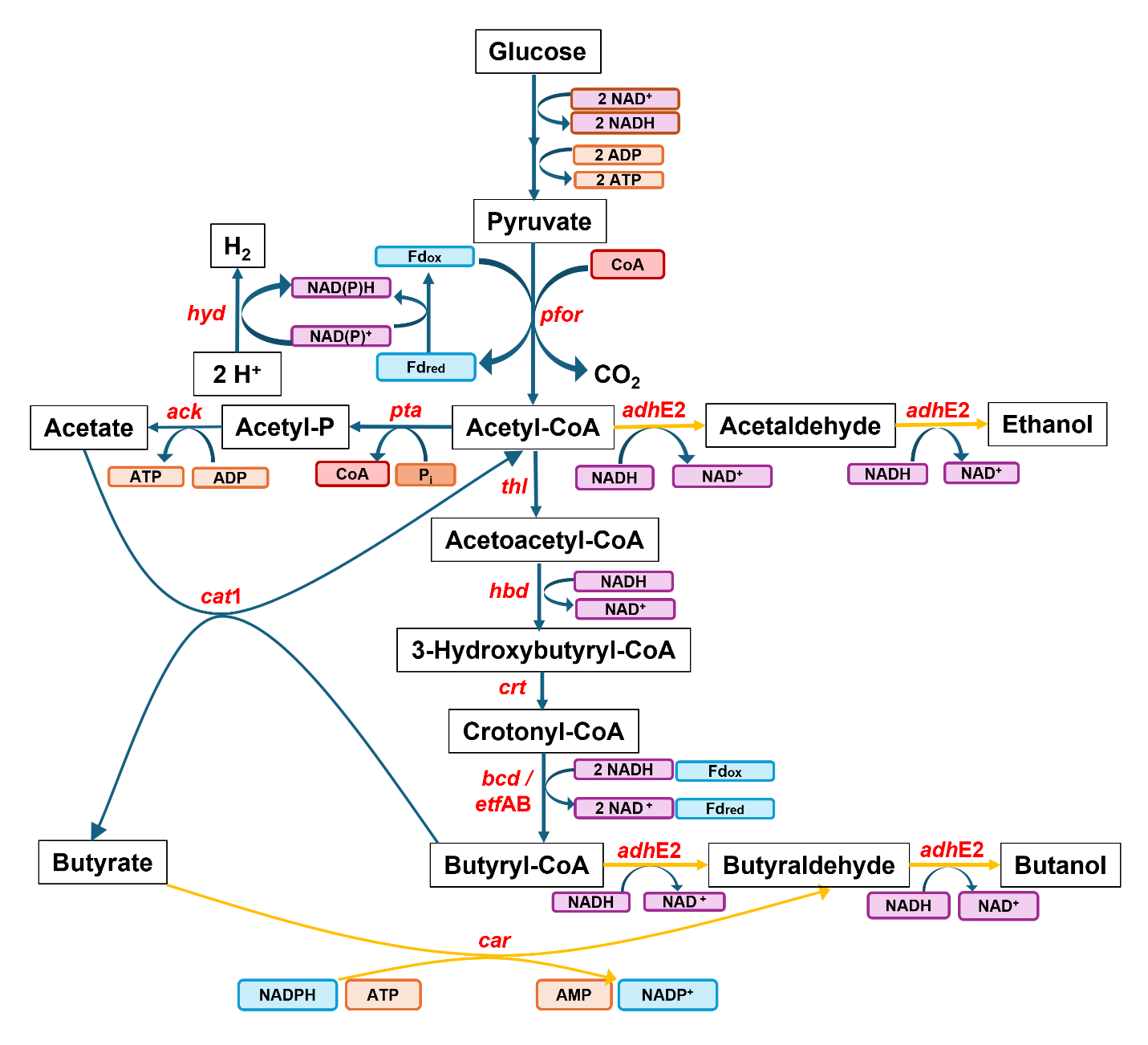
62]. The central metabolism for
C. tyrobutyricum engineered for n-butanol production is given in . Additionally,
C. tyrobutyricum does not contain the
ctfAB operon responsible for production of the acetoacetate intermediate. The absence of this operon essentially eliminates the possibility of acetone production.
. Metabolic pathways in engineered <em>C. tyrobutyricum</em> for butyrate and n-butanol production from glucose. The reactions catalyzed by the heterologous enzymes are in orange color. Genes in the pathways—<em>ack</em>: acetate kinase; <em>adh</em>E2: aldehyde/alcohol dehydrogenase; <em>adh </em>alcohol dehydrogenase; <em>bcd</em>: butyryl-CoA dehydrogenase; <em>cat</em>1: butyryl-CoA/acetate CoA transferase; <em>car</em>: carboxylic acid reductase; <em>crt</em>: crotonase; <em>etf</em>AB: electron transferring flavoprotein; <em>hbd</em>: 3-hydroxybutyryl-CoA dehydrogenase; <em>hyd</em>: hydrogenase; <em>pfor</em>: pyruvate:ferredoxin oxidoreductase; <em>pta</em>: phosphotransacetylase; <em>thl</em>: thiolase.
A major drawback to biobutanol production in
C. tyrobutyricum is its narrow substrate spectrum, which is capable of using limited monosaccharides for growth [
63].
C. tyrobutyricum’s comparatively smaller genome, when compared to other typical butanol-producing clostridia, lacks transporter and catabolism genes for the metabolism of more complex carbohydrates [
64]. Efforts have been made to increase the substrate variety of
C. tyrobutyricum, where heterologous catabolism pathways for various monosaccharides and disaccharides have been effectively implemented into
C. tyrobutyricum [
23,
24,
25].
2.4. Engineering Strategies for Increasing n-Butanol Production in C. tyrobutyricum
C. tyrobutyricum was first engineered to produce n-butanol in 2011 when Yu et al. overexpressed
adhE2 to reduce butyryl-CoA to butyraldehyde and eventually n-butanol [
18]. These researchers achieved a respectable yield of 0.27 g/g glucose. The butanol production in
C. tyrobutyricum was then increased with the generation of mutant strain Ack-
adhE2 (renamed from Δack-
adhE2), after optimizing the replicon in the conjugative plasmid and utilizing the more reduced substrate mannitol. This work resulted in n-butanol production at 20.5 g/L with a yield of 0.33 g/g [
65], yet this fermentation still suffered from high acid production. The group then addressed this by co-overexpressing CoA transferase (encoded by
ctfAB) from
C. acetobutylicum to increase n-butanol production via butyrate reassimilation. Butanol productivity and yield increased twofold [
22], but it was accompanied by the production of acetone. Zhang et al., 2018 used the native CRISPR-Cas system to knock out
cat1 with genomic insertion of
adhE2 in
C. tyrobutyricum, resulting in the mutant strain Δ
cat1::
adhE2 which produced 26.2 g/L n-butanol, with a yield of 0.23 g/g and negligible butyrate production from glucose [
26]. The fermentation with this mutant strain was still accompanied by significant acetate and ethanol production, which limited the butanol yield.
The production of n-butanol in engineered
C. tyrobutyricum overexpressing
adhE2 follows a very similar pathway to the one used in
C. acetobutylicum, where it was shown there is an additional need for NAD(P)H. To address this redox imbalance in
C. tyrobutyricum, researchers have taken a process engineering approach by supplementing methyl viologen (MV) as an electron carrier. This led to more than 80–90% reduction in acetic and butyric acids, increased n-butanol production to 14.5 g/L, and resulted in a yield greater than 0.3 g/g glucose in a
C. tyrobutyricum Ack-
adhE2 fermentation [
19]. This increase in butanol production was the result of suppressing hydrogen emissions and increasing NADH availability.
2.5. Metabolic Engineering of E. coli for n-Butanol Production
Escherichia coli is another microorganism that has been leveraged for biobutanol production [
66]. The Gram-negative, rod-shaped, facultative anaerobic
E. coli is well understood both physiologically and genetically.
E. coli can grow on minimal media, utilizes a variety of substrates as carbon sources, possesses a rapid growth rate, can handle a wide range of oxygen concentrations, is tolerant of industrial environments, and is resilient to elevated substrate and product levels, all factors making it an ideal cell factory [
67]. Much work has been done on engineering
E. coli to synthesize butanol. Comprehensive reviews on this topic can be found in recent review articles [
12,
66,
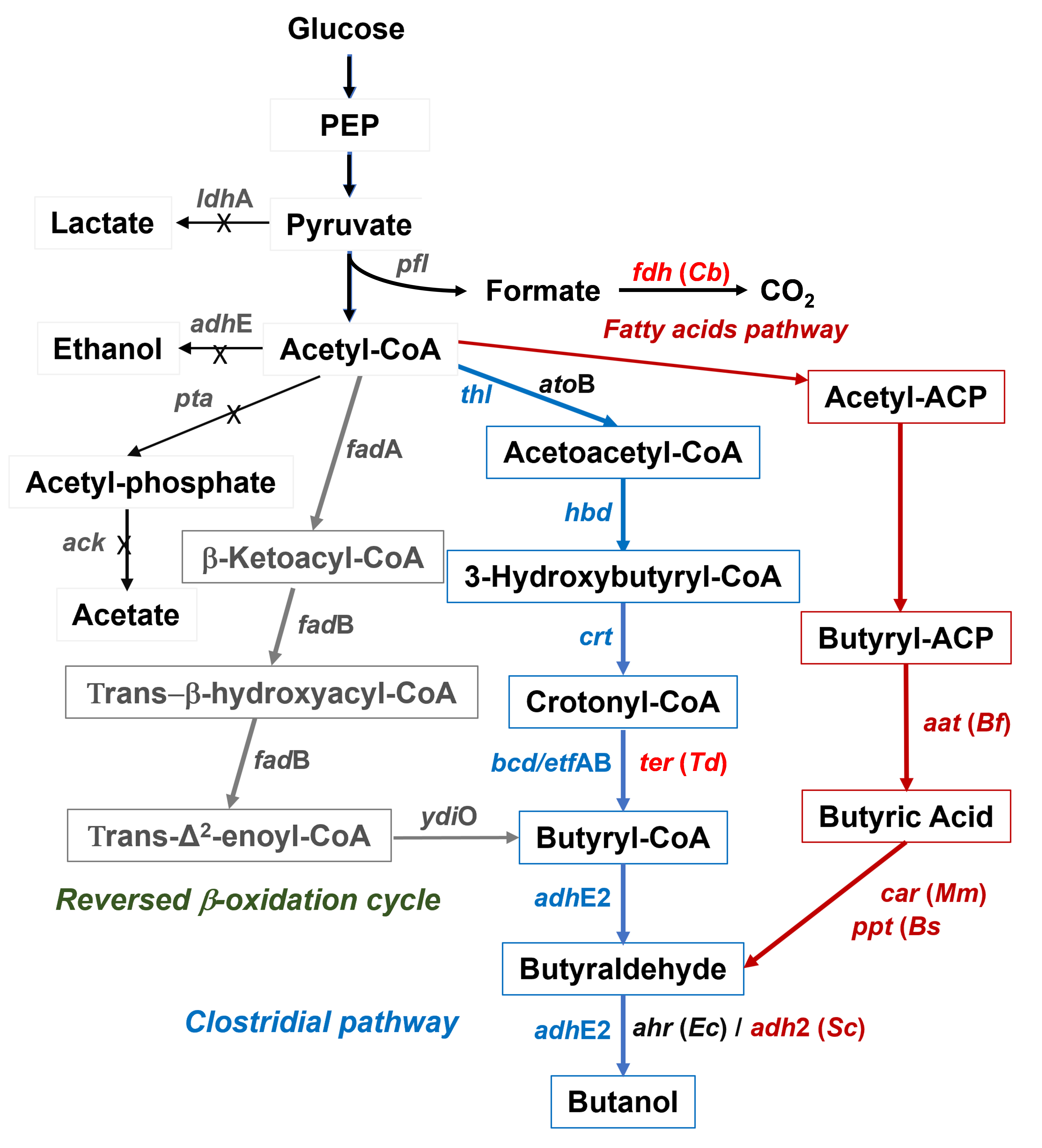
68]. A schematic overview of different metabolic engineering strategies used to produce n-butanol in
E. coli is given in .
. Overview of the different metabolic engineering strategies reported in the literature to produce n-butanol in <em>Escherichia coli</em>. <em>E. coli</em>’s native genes <em>ldh</em>A (lactate dehydrogenase), <em>adh</em>E (alcohol dehydrogenase), <em>ack</em> (acetate kinase), and <em>pta</em> (phosphotransacetylase), are knocked out to eliminate lactate, ethanol and acetate production, whereas a heterologous <em>fdh</em> (formate dehydrogenase) is overexpressed to convert formate produced from pyruvate via pyruvate formate lyase (<em>pfl</em>) to CO<sub>2</sub> to increase the intracellular NADH pool. The <em>Clostridial</em> n-butanol biosynthesis pathway with associated genes from <em>Clostridium acetobutylicum</em> is in blue. <em>ato</em>B (acetyl-CoA acetyltransferase) and <em>ter</em> (trans-2-enoyl-CoA reductase) are also used as better alternatives to <em>thl</em> and <em>bc</em>d/<em>etf</em>AB, respectively. The strategy is based on <em>E. coli</em>’s native fatty acids pathway and a heterologous pathway with associated genes <em>aat</em> (Acyl-ACP thioesterase),<em> car </em>(carboxylic acid reductase),<em> ppt</em> (phosphopantetheinyl transferase), and <em>ahr</em> or <em>adh</em>2 (aldehyde reductase) leading to butyric acid and then n-butanol is in red. The reversed β-oxidation cycle converting acetyl-CoA to butyryl-CoA via thiolase (<em>fad</em>A), hydroxyacyl-CoA dehydrogenase (<em>fad</em>B), enoyl-CoA hydratase (<em>fad</em>B), enoyl-CoA reductase (<em>ydi</em>O) is shown in grey. The sources of the genes are indicated in the parenthesis: <em>Bf</em>, <em>Bacteroides fragilis</em>; <em>Bs</em>, <em>Bacillus subtilis</em>; <em>Cb</em>, <em>Candida boidinii</em>; <em>Ec</em>, <em>Escherichia coli</em>; <em>Mm</em>, <em>Mycobacterium marinum</em>; <em>Td</em>, <em>Treponema denticola</em>. Abbreviation: ACP, acyl-acyl carrier protein.
Ethanol production was the first biofuel process explored in
E. coli, a compound it naturally produces [
69]. Atsumi et al., 2008 made a significant breakthrough by engineering
E. coli to produce n-butanol [
29]. This was made possible by introducing genes (
thl, crt, hbd, bcd-etfAB
, adhE) in the biosynthetic n-butanol pathway native to clostridia into the
E. coli genome. The study reported a butanol titer of 0.55 g/L by further replacing the thiolase (
thl) with the native
E. coli’s acetyl-CoA acetyltransferase (
atoB) and cultivating the cells semi-aerobically in nitrogen-rich media with 2% glycerol supplementation. Butanol production further increased to 15 g/L by replacing
bcd-etfAB with the
Treponema denticola’s
ter encoding trans-2-enoyl-CoA reductase that converted crotonyl-CoA to butyryl-CoA using NADH as a direct reducing equivalent without flavoproteins or ferredoxin [
30]. The engineered
E. coli strain also had its native
pta encoding phosphate acetyltransferase knockout to direct the acetyl-CoA toward n-butanol, instead of acetate, biosynthesis. In addition, formate dehydrogenase (
fdh) from
Candida boidinii was overexpressed to oxidize formate to CO
2 and increase the intracellular NADH driving force, resulting in increased butanol production from glucose at a high yield of ~0.28 g/g or 70% of theoretical in fed-batch fermentation with
in-situ butanol separation by gas stripping.
Despite the low initial butanol titers, there have been major advancements in n-butanol production in
E. coli. For example, Dong et al., 2017 developed an
E. coli strain by completely engineering the chromosome [
31]. It could produce butanol without the need of plasmids, antibiotics, or inducing agents. First, the butanol pathway was inserted into the chromosome, and then the host and the butanol pathway were engineered iteratively to stabilize the heterologous butanol pathway in the host. This resulted in
E. coli strain EB243, with 33 native genes deletions and 5 heterologous genes expressed. The fermentation of strain EB243 gave a titer of 20 g/L butanol, with a yield reaching 83% of the theoretical yield in batch fermentation. This work is an example of how far butanol production in
E. coli has come.
An earlier work also demonstrated n-butanol production from glucose in
E. coli with engineered reversal of the β-oxidation cycle involving thiolase (
yqeF,
fadA), hydroxyacyl-CoA dehydrogenase (
fadB), enoyl-CoA hydratase (
fadB), enoyl-CoA reductase (
ydiO), and alcohol dehydrogenases, achieving a final butanol titer of 13.5 g/L, a yield of 0.27 g/g, and productivity of 0.33 g/L·h in batch fermentation with aeration controlled at 5% saturation of the dissolved oxygen [
33].
Most
E. coli n-butanol production studies focused on engineering the CoA-dependent clostridial pathway, which was anaerobic and required complex nutrients for cell growth. Leveraging the native fatty acids biosynthesis pathway in
E. coli, an innovative, O
2-tolerant butanol biosynthesis pathway has been constructed in
E. coli by expressing Acyl-ACP thioesterase (AAT) specific to butyryl-ACP and an oxygen-tolerant carboxylic acid reductase (CAR) [
70]. The AAT catalyzed butyric acid production from butyryl-ACP, CAR catalyzed butyraldehyde production from the generated butyric acid, and an
E. coli aldehyde reductase (
ahr) then catalyzed the production of n-butanol from butyraldehyde, resulting in butanol production of ~0.3 g/L. More recently, an
E. coli strain overexpressing CAR from
Mycobacterium marinum, phosphopantetheinyl transferase (PPT encoded by
sfp) from
Bacillus subtilis, alcohol dehydrogenase (
adh2) from
Saccharomyces cerevisiae and the thioesterase TesBT was constructed to produce n-butanol from glucose, reaching ~2.9 g/L in a fed-batch fermentation [
32]. Although the butanol production via the fatty acids pathway was quite low compared to the clostridial pathway, the novel development here was the use of the heterologous CAR enzyme for the reduction of butyric acid to butyraldehyde. This typically recalcitrant reaction is easily catalyzed by the CAR enzyme.
3. Carboxylic Acid Reductases in Butanol Biosynthesis
3.1. Function and Mechanism of Carboxylic Acid Reductases
Carboxylic acid reductases (CARs) catalyze the reduction of carboxylic acids into their corresponding aldehydes. Classified under EC 1.2.1.30, CARs work by first activating the carboxylate substrate using ATP, then reduce the carboxylate substrate with NADPH serving as the hydride donor [
71]. The reaction is given in the equation below.
This metabolic activity was first witnessed in fungi [
72], though it was not until the late 1960s that the CAR protein native to the fungus
Neurospora crassa (NcCAR) was purified and characterized [
73]. More recently, there has been a surge in interest in CARs.
CARs are promiscuous enzymes, characterized by a broad substrate range. The substrate specificities largely overlap across CARs with some exceptions, like SrCAR’s unique ability to reduce a nitro-substituted benzoic acid derivative [
74]. CARs were originally referred to as aryl-aldehyde dehydrogenases (NADP
+), reflecting their preference for aromatic substrates. Now, it is universally known that CARs are also effective at reducing short to intermediate chain length aliphatic acids.
CARs are composed of three domains: an adenylation domain (A-domain), a transthiolation domain (T-domain), and a reductase domain (R-domain). CARs undergo a post-translational modification, where a conserved serine in the T-domain is activated by a phosphopantetheinyl transferase. The CAR catalytic cycle begins with the activation of the carboxylate substrate. The exact mechanism of aldehyde formation by CAR remains unproven, but current understanding suggests that the process begins when ATP activates the carboxylic acid substrate, creating an AMP-ester. This step is followed by the thiol group of phosphopantetheine catalyzing the transesterification of the AMP-ester, forming a thioester that covalently attaches the acyl group to the CAR. The substrate is then transported from the A-domain to the R-domain, via the T-domain in a “swinging arm” motion [
75]. The final step is the reduction of the thioester in the R-domain by NADPH [
76]. Although these steps are consistent with the current understanding of the process, further experimental validation is needed, potentially through mutational and kinetic studies like those performed for NRPS domain modifications. DNA alignment of empirically verified CARs has elucidated a CAR signature, composed of four distinct segments containing conserved residues within the A-domain., the phosphopantetheinylated serine region in the T-domain, and three conserved regions in the R-domain. This information is crucial for identifying new CAR sequences.
Researchers have a keen interest in CAR regarding n-butanol production because it can be used to reassimilate by-products for further n-butanol production, and it can be used to engineer novel n-butanol producing pathways. CAR use has been successfully demonstrated in
E. coli for n-butanol production [
32]. As discussed earlier, activity on the substate butyrate by CAR, isolated from
Mycobacterium marinum (mmCAR), has already been demonstrated in
E. coli co-expressing mmCAR,
sfp, and
adh2. Until recently, CAR had not yet been utilized in n-butanol production occurring in a clostridia species. There is high potential for its use since it has been demonstrated that it can reduce butyric acid to butyraldehyde. Reduction to butyraldehyde via CAR could increase n-butanol production in
C. tyrobutyricum since the major byproduct of this n-butanol fermentation is butyric acid. Our most recent study showed that overexpressing mmCAR and
sfp in the
C. tyrobutyricum strain overexpressing
adhE2 increased butanol production with reduced butyrate production compared to the strain without co-expressing mmCAR and
sfp [
77].
The promise of selective aldehyde production drives CAR research. However, cofactor demand is a major limitation of CAR utilization in bioproduction.
In vitro, the high cost of cofactors is inhibitory, and the use of CAR creates a balance between cofactor supply and product selectivity, as the cellular environment reactions often eliminate reactive aldehyde species. To enhance
in-vivo cascades, one effective approach is to adjust the aldehyde levels to lower concentrations. This can be achieved by fine-tuning the reaction rates of the subsequent transformations. Another method is through channeling, examples include scaffolding proteins or utilizing microcompartments [
78].
3.2. Engineering Efforts to Enhance CAR Enzyme Performance
Although some steps of the CAR mechanism remain unclear, there is enough information to achieve kinetic improvements via rational engineering. In work performed by Wang et al., 2022, the catalytic efficiency of a
Mycobacterium smegmatis originating CAR was increased by engineering the “hinge” mechanism of the CAR enzyme [
79]. The team designed a mutagenesis library of the hinge region based on the characteristic construction of CARs and their superfamily. The team was able to achieve a 6.57-fold improvement in catalytic efficiency by introducing the mutations R505I/N506K. The increase in catalytic efficiency is attributable to tight binding of the acyl-AMP complex, as explained by MD simulations.
4. Cofactor Regeneration Systems
4.1. Cofactor Regeneration Overview
NADH availability is a limiting factor in butanol-producing solventogenic clostridia [
80]. Strategies employed to overcome this challenge typically fall into one of two categories. The first strategy is metabolic or redox engineering, which involves altering the organism’s flux distributions to restore redox balance. The second strategy, known as “Metabolic Process Engineering” (MPE), adjusts the fluxes within the metabolic pathway by manipulating the fermentation environment, without requiring genetic modifications to the organism. For example, the availability of NADH and the resulting butanol production were notably improved when glycerol and mannitol,
i.e., more reduced substrates, were used for fermentation [
18,
81]. Another potential example of MPE that can be utilized is cofactor regeneration.
Cofactors act as enzyme reaction agents, interacting directly with substrates, typically changing only in oxidation state as a driving force for chemical reactions. However, direct stoichiometric supplementation of cofactors in industrial applications is cost-prohibitive. Therefore, there is an essential need to regenerate cofactors. The regeneration of nicotinamide cofactors can be performed using a variety of methods, including enzymatic, chemical, photochemical, and electrochemical approaches [
82]. Enzymatic cofactor regeneration typically occurs via one of two methods. The first method employs a single enzyme, utilizing both the reduced and oxidized forms of the cofactor, coupling the synthesis of the desired product with the cofactor regeneration reaction. The second method uses two separate enzymes, one for product synthesis and the other for cofactor regeneration. Yoo et al., 2017 described the attributes of an effective regeneration system, which must meet the following criteria: (1) the process should be practical and cost-effective, (2) the regeneration system must be stable, (3) the separation of the product should be straightforward, and (4) byproduct formation should be minimal [
83]. shows some examples of cofactor regeneration using various enzymes, including formate dehydrogenase (FDH), glucose dehydrogenase (GDH), and phosphite dehydrogenase (PTDH) in notable biotransformation studies. To date, only FDH and PTDH have been investigated for their potential to increase intracellular NADH levels and butanol production in clostridia.
.
Examples of cofactor regeneration and the enzymes used in notable biotransformation studies.
| Cofactor |
Cofactor Consuming Enzyme |
Cofactor Regenerating Enzyme |
Application |
Reference |
| NADH |
L-PheDH |
NADH oxidase |
NADH oxidase from Lactobacillus brevis was used to regenerate NAD+ to increase L-methionine conversion.
RESULT: L-Methionine conversion of 100% was achieved in the batch system using NADH oxidase as the regeneration enzyme.
|
[84] |
| NADH |
LDH |
FDH |
D-lactate dehydrogenase (LDH) was used to transform 2-oxo-4-phenylbutyric acid (OPBA) to R-HPBA, with concomitant oxidation of NADH to NAD+, which was regenerated by formate dehydrogenase (FDH) present in whole cells of Candida boidinii.
RESULT: R-HPBA production with yeast cells and LDH co-immobilized in a fibrous bed bioreactor operated in fed-batch and repeated batch modes gave stable productivity of 9 g/L·h and yield of 0.95 mol/mol OPBA.
|
[85] |
| NADPH |
AKR, CAR |
FDH |
In-vivo and in-vitro enzyme cascades were used to generate 1,4-butanediol and 1,6-hexanediol from 4-hydroxybutanoic and adipic acids. Mutated FDH was utilized for NADPH regeneration.
RESULT: in-vitro biotransformations resulted in 90% conversion of 4-HB to 1,4-BDO, and 76% conversion of adipic acid to 1,6-HDO. In-vivo biotransformations resulted in >95% substrate conversion for both 4-HB and adipic acids.
|
[86] |
| NADPH |
DAPDH |
FDH |
In-vivo biocatalysis system for the synthesis of D‑amino acids from L‑amino acids by the co‑expression of membrane‑associated L‑amino acid deaminase from Proteus mirabilis, meso‑diaminopimelate dehydrogenases from Symbiobacterium thermophilum, and formate dehydrogenase from Burkholderia stabilis, in recombinant E. coli.
RESULT: E. coli whole‑cell biocatalyst asymmetrically catalyzed the stereoinversion of L‑Phe to D‑Phe, with yields >99% in 24 h.
|
[87] |
| NADPH |
SDH |
PTDH |
Ralstonia sp. 4506 PTDH was mutated to confer higher selectivity toward NADPH and was utilized in the biotransformation of 3-dehydroshikimate to shikimic acid.
RESULT: Thermostable and selective PTDH mutant RsPtxDHARRA showed >95% substrate conversion after 90 min at 45 °C, whereas wild-type RsPtxD reaction ceased after 30 min.
|
[88] |
| NCD |
D-LDH |
FDH |
FDH from Pseudomonas sp. 101 was engineered to regenerate nicotinamide cytosine dinucleotide (NCD) selectively.
RESULT: Most active mutants reached a cofactor preference switch from NAD to NCD by 3700-fold. FDH was coupled with NCD-dependent D-lactate dehydrogenase, where 5.0 mM formate was exhausted in 30 min with an equal amount of D-lactate generation, displaying a perfect stoichiometry.
|
[89] |
| NMNH |
multiple |
GDH |
In-vitro transformations utilizing a GDH engineered for NMNH specificity and NMNH supplementation, combined with one of three different reductases: XenA, OYE3, or NfsB. In-vivo biotransformations with multiple reductases co-expressed simultaneously along with a GDH for cofactor regeneration.
RESULT: Transformations using these enzyme combinations with NMNH supplementations resulted in conversions of >99%, >99%, and >92% on their respective substrates. In-vivo biotransformations showed the ability to selectively produce levodione based on the GDH mutant, due to XenA and GDH specificity toward NMNH.
|
[90] |
| ATP |
CAR |
PPK12 |
PPK12 was evaluated for ATP recycling in car-catalyzed 4-methoxybenzoic acid reduction.
RESULT: reaction reached >99% conversion required 20% less substrate than the control and half the duration
|
[91] |
4.2. Formate Dehydrogenase
Formate dehydrogenase (FDH, E.C. 1.2.1.2) catalyzes the oxidation of formate to carbon dioxide by reducing NAD
+ to NADH [
92]. The innocuous carbon dioxide produced can be released safely into the environment. Formate is inexpensive and non-toxic to most enzymes, serving as a suitable substrate. The irreversibility of this reaction offers a significant advantage for commercial applications. Formate dehydrogenase is the most developed enzyme for cofactor regeneration and is commercially available. Several studies demonstrate the utility of FDH in
Clostridium. For instance, the NAD
+-dependent formate dehydrogenase FDH1 gene from
Candida boidinii was overexpressed for NADH regeneration in
Clostridium paraputrificum, which increased hydrogen yield by 59% compared with the control [
93]. Similarly, the expression of the heterologous formate dehydrogenase in
Clostridium ljungdahlii also facilitated the regeneration of NADH and increased its intracellular concentration from 2.93 to 12.64 μM per g DCW [
94]. In
Clostridium tyrobutyricum, introducing a heterologous formate dehydrogenase with the bifunctional aldehyde/alcohol dehydrogenase increased butanol production from glucose to 12.3 g/L, an increase of ~18% compared to the control strain without expressing the FDH [
95].
4.3. Glucose Dehydrogenase
Glucose dehydrogenase is another enzyme being developed for cofactor regeneration. GDH (E.C. 1.1.1.47) catalyzes the β-
d-glucose to β-
d-glucono-1,5-lactone oxidation reaction while concurrently reduces the cofactor NAD(P)
+ to NAD(P)H. However, it also catalyzes NAD
+ to NADH to a lesser extent [
96]. There are several examples of GDH used for cofactor regeneration. For example, the GDH from
Bacillus sp. [
97] was used to regenerate NADH in the synthesis of L-6-hydroxynorleucine from 2-keto-6-hydroxyhexanoic acid via glutamate dehydrogenase. GDH from
Bacillus sp. was again used in NADH regeneration in the production of L-carnitine from 3-dehydrocarnitine via L-carnitine dehydrogenase [
98].
4.4. Phosphite Dehydrogenase
A third, and more recently discovered enzyme used in cofactor regeneration is phosphite dehydrogenase (PTDH, E.C. 1.20.1.1), an NAD
+ dependent enzyme oxidizing inorganic phosphite (hydrogen phosphonate) to phosphate [
99,
100]. This reaction is given in the equation below [
101]:
This reaction is unique in that there is a hydride donor to hydroxide acceptor phosphoryl transfer, a rare occurrence. During the phosphorus atom oxidation process, the phosphorus atom’s oxidation state changes from +3 to +5, resulting in a -0.65V redox potential for the phosphite/phosphate couple. This makes the phosphite oxidation reaction essentially irreversible, a major advantage of using PTDH for cofactor regeneration.
The most extensively studied PTDH enzyme originates from the microorganism
Pseudomonas stutzeri WM88. This enzyme was first described by White and Metcalf, 2002 to be effective at oxidizing reduced phosphorus compounds [
100]. This strain was initially detected by using a selection medium consisting solely of hypophosphite or phosphite as the only phosphorus sources. It was revealed that hypophosphite is oxidized to phosphate in a two-step process, with phosphite as an intermediate. The
htx and
ptx loci are the responsible gene clusters, with the gene product PtxD (PTDH) being the agent responsible for catalyzing the phosphite oxidation [
99]. The expression of the PtxD gene could be induced by phosphate starvation [
102].
The PTDH enzyme shares a resemblance to D-hydroxy acid dehydrogenases, while reactions catalyzed by PTDH are notably different. It shares the key active site residues, including Arg237, Glu266, and His292 required for binding and/or catalysis [
101]. His292 is likely the active site base responsible for deprotonating the water nucleophile, though it is possible that it serves as a nucleophilic catalyst. Other critical residues and their role include: Arg237 (phosphite binding), Glu266 (positioning of residues His292 and Arg237), Lys76 (stabilization of the dianionic phosphite substrate), and Glu175 and Ala176 (cofactor binding and cofactor specificity). It is suspected that the rate limiting step is the hydride transfer from the phosphite to the nicotinamide cofactor [
103]. PTDH has a strict specificity for NAD
+, however, using rational design methods, it has been engineered to reduce NADP
+. Using site directed mutagenesis researchers were able to eliminate steric and repulsive constraints for NADP binding [
104]. Replacing the two residues Glu175 and Ala176 with Ala175 and Arg176, respectively, produced PTDH mutants with enhanced cofactor promiscuity. All mutants created demonstrated drastically improved catalytic efficiency for both cofactors, with the double mutant showing the best kinetic performance. The G175A/A176R mutant exhibited a 3.6-fold increase in catalytic efficiency for NAD
+ and a 1000-fold increase for NADP
+. The preference shifted from a 100-fold bias toward NAD
+ in the wild-type enzyme to a 3-fold preference for NADP
+ in the double mutant. Co-expressing the mutated PTDH with
adhE2 in
C. tyrobutyricum showed dramatically increased butanol and reduced butyrate and acetate production compared to the strain without co-expressing the PTDH [
77].
5. Protein Engineering of Aldehyde/Alcohol Dehydrogenase for Enhanced Butanol Production
5.1. AAD Overview
Many studies have focused on metabolic engineering to increase n-butanol production, whereas utilizing protein engineering to increase the metabolic flux of a particular reaction is typically an afterthought. Various studies have started to consider this, with appealing results. Often, good enzyme candidates for protein engineering have high substrate promiscuity, low turnover, or strict selectivity for a limited or unavailable cofactor. One enzyme in the
C. acetobutylicum metabolic pathway that checks many of these boxes is the aldehyde/alcohol dehydrogenase (AAD) encoded by
adhE2.
In continuous culture,
C. acetobutylicum is typically characterized as having two distinct metabolic states [
105], acidogenesis (production of acetate and butyrate) when grown near pH seven on glucose, and solventogenesis (production of acetone, butanol, and ethanol) when grown at low pH on glucose. However, a third, distinct, metabolic state exists; alcohologenesis (formation of butanol and ethanol but not acetone) occurs when grown at near pH seven under conditions of high nicotinamide cofactor availability. It was discovered that in methyl viologen induced alcohologenic cultures, the genes involved in solventogenesis for butanol biosynthesis were not transcribed, leading to the discovery of
adhE2 [
106].
AdhE2 encodes a bifunctional, aldehyde/alcohol dehydrogenase with strict NADH dependence responsible for butanol production in alcohologenic
C. acetobutylicum cultures. The aldehyde dehydrogenase (ALDH) domain is located at the N terminal of the gene, where a conserved region is located as characteristic of an aldehyde dehydrogenase catalytic center [
107]. The domain also contains the two nucleotide binding sites, GVVXG and GCGXXG, sites conserved in ALDH proteins [
106]. The alcohol dehydrogenase (ADH) domain is located at the C terminal of the gene, where it possesses a GXGXXVXXA conserved sequence, involved in coenzyme binding. The ADH domain also contains the three histidines of the iron-binding motif, a highly conserved iron-binding site.
A recent study [
108] elucidated the roles of two AADs,
adhE1 and
adhE2, in the primary metabolism of
C. acetobutylicum. The study revealed that, regarding n-butanol formation,
adhE1 plays the prominent role during solventogenesis, whereas
adhE2 plays a prominent role during acidogenesis and alcohologenesis.
Little research has been done on the specific structure and catalytic mechanism of the
C. acetobutylicum adhE2 enzyme, but it shares much sequence similarity with the
E. coli adhE gene. We can infer features of the
adhE2 enzyme based on this similarity. The
adhE enzyme from
E. coli is a bifunctional aldehyde/alcohol dehydrogenase highly conserved in a variety of organisms, with
adhE playing a major role in anaerobic conditions. The reduction of acetyl-CoA to ethanol is a two-step reaction: acetyl-CoA is converted to acetaldehyde in the N-terminal aldehyde dehydrogenase (ALDH) domain, and acetaldehyde is converted to ethanol in the C-terminal alcohol dehydrogenase (ADH) domain, with both domains linked together through a short linker. The close proximity of these two domains to each other likely indicates there is substrate channeling to improve the ethanol production rate [
109].
5.2. Engineering AADs for Improved Catalytic Efficiency
Several studies have implied that the final steps in the metabolic pathway of n-butanol production, butyryl-CoA reduction and butyraldehyde reduction, can be rate limiting. Increasing the turnover of the
adhE2 enzyme can relieve this bottleneck and increase butanol production. Increasing butanol selectivity is also on the wish-list. These mutation studies have not yet been performed on
adhE2, but studies have been done on similar enzymes, such as
adhE1 from
C. acetobutylicum and alcohol dehydrogenase from
Zymomonas mobilis. Rellos et al., 1997 studied the substrate specificity of
Z. mobilis alcohol dehydrogenase-2 enzyme using random mutagenesis and discovered that mutations to residues 161, and 155 and 165 enabled the enzyme to accommodate longer-chain alcohols [
110]. Specifically, the researchers created the double point mutant A161I, R165H with a V
max that was 135-fold higher than the wild type. This study is a demonstration of the ability to increase enzyme turnover by active-site mutation. Another great example of increasing activity through mutation is the work performed by Cho et al., 2019, who used site-saturation mutagenesis to mutate residues close to the active site of the alcohol dehydrogenase domain of
adhE1 from
C. acetobutylicum [
111]. In their work, two mutations, F716L and N655H, were found to drastically increase the butanol: ethanol ratios by 5.8-fold and 5.3-fold, respectively, when compared to the wild-type enzyme. lists some of the notable studies in engineering AAD for enhanced butanol production.
.
Notable examples of engineering alcohol dehydrogenase to increase production.
| Dehydrogenase Enzyme |
Mutation(s) |
Description |
Reference |
| ZADH2 |
A161I, R165H; D38G |
Mutations to residues 161 and 165 increase activity with longer-chain alcohols, mutations to residue 38 increases NADPH activity |
[110] |
| AdhE1-AAD |
M619G, N655H, F716L |
mutations increased the butanol:ethanol ratio from fermentation for C. acetobutylicum M5 strain by 2.22-, 5.3-, and 5.8-fold |
[111] |
| AdhE1-AAD |
D485G |
The D485G mutation increased butanol yield by 4% |
[46] |
| AdhE-AAD |
D494G |
D494G mutation made to C. thermocellum adhE-AAD. Used to increase NADPH cofactor specificity. Fermentation using this AAD resulted in 1.7-fold increase in butanol yield when compared to the wild-type strain. |
[112] |
| AdhD |
G211C, G211InsC |
Mutations were made to the AdhD gene from Pyrococcus furiosus to increase NADPH activity. Both mutations, the G211C point mutation and the Cysteine insertion, resulted in an activity increase of over 90-fold. |
[113] |
| TbSADH |
H42T |
The Thermoanaerobacter brockii ADH was engineered to improve the proton transfer process. The H42T mutant showed a >99% conversion. |
[114] |
6. Challenges and Future Directions
6.1. Challenges Associated with AAD Engineering
The metabolic and protein design strategies described above are vastly different, and each one has its own challenges and limitations. Protein engineering of the AAD enzyme by either rational design or directed evolution strategies shows great promise for increasing n-butanol production. One challenge that has become apparent is that as the enzyme is mutated to increase the turnover of the larger butyraldehyde substrate, it typically also increases acetaldehyde turnover. What is sought is the increase in butyraldehyde turnover and a decrease in acetaldehyde turnover to increase the selectivity of the enzyme meanwhile maintaining or increasing the catalytic efficiency. This is a classic “multi-state” design problem. The use of directed evolution in this case is straightforward, one directs the evolution of this enzyme to increase butanol production meanwhile maintaining or increasing selectivity. In the past, multi-state design problems were challenging to address when using rational design approaches. There are plenty of examples in the literature where rational design has been used to address multi-state design problems with good results. For example, Schmitz et al., 2017 developed a multi-state framework (MSF) in the Rosetta Macromolecular Modeling Suite that enables the implementation of Rosetta’s single-state protocols in a multi-state environment [
115]. Utilizing MSF, the researchers demonstrated a 15% higher performance using MSF than single-state design on a ligand-binding benchmark. The researchers then used this protocol to design nine
de-novo, retro-aldolases, with all variants displaying measurable catalytic activity, thus illustrating the efficacy of the concept. By utilizing these new design tools to engineer AAD for increased selectivity, it may be possible to achieve higher butanol titers and yields than using single-state rational design alone.
6.2. Challenges Associated with PTDH Usage
An additional challenge, this time presented from PTDH use for cofactor regeneration, is the fact that a general increase in the redox ratio of nicotinamide cofactors may be detrimental to bioproduction. This could be due to cofactor transcriptional regulation, overproduction of side-products, or substrate inhibition of an accompanying enzyme in the cascade. There has been work undertaken to address these limitations, and the answer may be the use of orthogonal cofactors. Orthogonal cofactors, also known as noncanonical cofactor biomimetics (NCBs), are moieties that can function as cofactors in enzymatic catalysis yet are structurally distinguishable from natural nicotinamide cofactors [
116]. The use of NCBs offers significant advantages, including improved stability [
117], and the ability to reduce interference between modified pathways and the host’s natural metabolic processes [
90,
118]. A commonly used NCB is nicotinamide mononucleotide NMN(H). NMN(H) is identical to the nicotinamide cofactors but lacks the adenosine binding handle used in enzyme recognition. Black et al., 2020 developed a glucose dehydrogenase with a 107-fold shift in cofactor specificity favoring NMN
+ over NADP
+ [
90]. The researchers then demonstrated the ability of the system to drive diverse redox reactions
in vitro, with a high total turnover number (~39,000). Finally, the toolkit was completed by establishing Nox Ortho [
119], a reduced NMN
+(NMNH)-specific oxidase. When GDH ortho and Nox ortho are used together the enzymes can modulate NMNH:NMN
+ ratio. The team showed the NMN(H) cofactor pool could be manipulated to achieve a chosen redox ratio at will, ranging from 0.07 to 70 for NMNH:NMN
+. Additionally, this NMN(H) cofactor pool is dissociated from both nicotinamide cofactor redox ratios. A (S)-specific butanediol (BDO) dehydrogenase was then designed to utilize NMN(H). Using the orthogonal, NMN(H) driving force, chiral-pure 2,3-BDO was produced to near completion, unaffected by NAD(P)/H reduction potentials
in vitro or
in vivo.
6.3. Challenges Associated with CAR Usage
For CAR, a major limitation of the use of this enzyme is its need for NADPH and ATP cofactor. Again, it may be possible to address both using cofactor regeneration, although the regeneration of cofactor ATP has not been as straightforward. ATP regenerating methods starting from ADP are well-established, but these methods have disadvantages such as price, instability, and phosphate donor availability [
120]. For these reasons more attention has been given to the polyphosphate kinase (PPK)/polyphosphate (polyP) ATP regeneration system. It was thought that there were only two PPK2 classes, PPK2-I and PPK2-II, where PPK2-II would generate nucleoside 5′-diphosphates from nucleoside 5′-monophosphates and polyphosphate consumption and PPK2-I would generate nucleoside 5′-triphosphates from the corresponding nucleoside 5′-diphosphates and polyphosphate consumption. But the recently discovered PPK2-III shows great promise because it is bifunctional, capable of forming both nucleoside-5′ diphosphates and nucleoside 5′-triphosphates, reducing the need to overexpress two separate PPK2 enzymes for ATP regeneration [
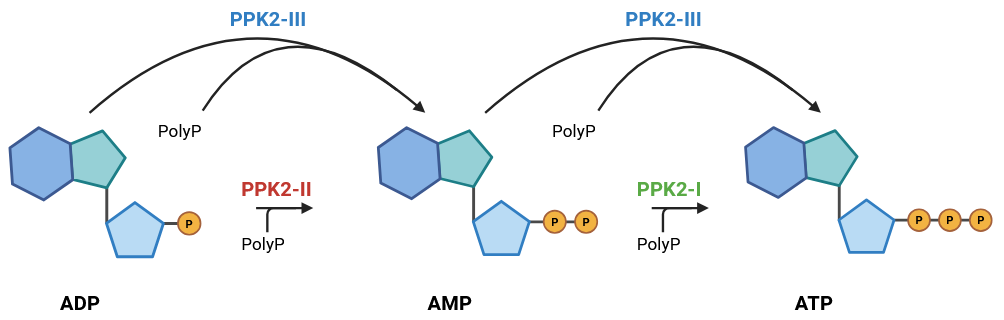
121]. The PPK2 subfamilies and preferred catalyzed reaction(s) related to ATP regeneration are given in . The promise of the PPK2-III class of enzyme is highlighted in the work performed by Tavanti et al., who screened a panel of 82 putative PPK2-III enzymes to discover the gene PPK12, isolated from an unclassified
Erysipelotrichaceae bacterium [
91]. The researchers used the PPK12 enzyme for polyP-based ATP recycling to power aldehyde production via a CAR catalyzed reaction. This resulted in several gram scales, cell free aldehyde synthesis, needing substantially less enzyme compared to the ajPAP process, a PPK2-II enzyme of high promise. This work emphasizes the feasibility of ATP regeneration using a PPK2-II enzyme for carboxylic acid reduction via CAR.
. PPK2 subfamilies and preferred catalyzed reaction(s) related to ATP regeneration.
It is noted that ferredoxin-associated aldehyde oxidoreductase (AOR) can also reduce carboxylic acids to aldehydes under anaerobic conditions. Autotrophic acetogens such as
C. ljungdahlii and
C. autoethanogenum produce ethanol from acetate via AOR by reducing acetate to acetaldehyde and then to ethanol via ADH [
122,
123].
C. carboxidivorans produces ethanol and n-butanol via the AOR-ADH pathway under autotrophic growth and the ALD-ADH pathway under heterotrophic conditions. AOR plays a critical role in ATP generation under energy-limited conditions and ethanol/acetate formation in acetogens during gas fermentation [
124]. Also, exogenous acetate can promote the expression of AOR and ethanol production from acetate [
125]. However, to maintain redox and energy balance in response to different fermentation conditions, AOR catalyzes the reaction from acetate to acetaldehyde in CO fermentation but catalyzes the reverse reaction in CO
2/H
2 fermentation [
126]. AOR’s function also depends on pH, reducing acetate/butyrate to aldehydes with reduced ferredoxin at pH 4.0–5.0 while oxidizing aldehydes to acetate/butyrate with oxidized ferredoxin at pH >5.0 [
127]. Overexpressing
aor or
adhE2 in
C. carboxidivorans increased its alcohol production from glucose, demonstrating the potential of increasing alcohol production by overexpressing these genes in two different alcohol production pathways [
128]. An earlier study has also shown that the resting cells of
C. formicoaceticum converted carboxylates to corresponding alcohols in the presence of CO or formate and an electron mediator [
129], confirming the presence of ALD, ADH, and AOR activities in
C. formicoaceticum and their ability to convert acetate to ethanol and butyrate to n-butanol [
127]. However, AOR uses reduced ferredoxin (Fd
red) as the cofactor, which has a low redox potential (ca. −420 mV at pH 7.0), in transferring the electron to carboxylate. The regeneration of Fd
red requires a highly reduced electron donor, such as the reduced methyl viologen (E
h = −450 mV), which may be too expensive to use in industrial fermentation.
7. Conclusions and Prospects
Biobutanol production is limited by available reducing equivalents (NADH, NADPH, and Fd
red) and the coproduction of ethanol, acetate, and butyrate. Metabolic and redox engineering approaches have been used to increase butanol production, with the butanol yield exceeding 80% of the theoretical yield (0.41 g/g glucose). However, about one third of the substrate carbon in glucose is lost as CO
2 in glycolysis through the EMP pathway, which not only limits product yield but also causes environmental concern due to the release of CO
2, a major greenhouse gas. It is thus desirable to capture and utilize the fermentation-produced gases (CO
2 and H
2) for butanol production. There are carboxydotrophic acetogens that can convert CO
2 to acetyl-CoA with H
2 or CO as the energy source and electron donor via the Wood–Ljungdahl pathway [
130]. Some mixotrophic acetogens can concurrently utilize sugars and gases (CO
2 and H
2) for acetate production with a >95% (
w/
w) yield without losing much carbon to cell biomass or CO
2 [
20,
131,
132,
133]. However, gas fermentation is limited by the extremely low solubility of CO
2 and H
2 in water. To overcome this problem, CO
2 can be converted to formate first via an electrochemical reduction process [
134] and then to acetate using a homoacetogen such as
C. formicoaceticum. Since
C. tyrobutyricum can convert glucose and acetate as co-substrates to butyrate [
135], it is possible to convert all glucose carbon to butyrate by reassimilating the CO
2 released in glycolysis through formate and acetate by using
C. formicoaceticum. If
C. tyrobutyricum expressing
adhE2 can be further engineered to efficiently utilize CAR or AOR to convert butyrate to butyraldehyde and then to butanol, it is possible to attain an overall butanol yield of 0.6 g/g glucose without any CO
2 emission. This may require the use of orthogonal cofactors such as NMN(H) to provide additional reducing equivalents needed for butanol biosynthesis. Together with the engineered AAD (encoded by the
adhE2) with a high selectivity for butanol over ethanol, a novel butanol producing strain with close to 100% carbon recovery can be developed for sustainable production of biobutanol for fuel and industrial applications.
Author Contributions
C.D.M.: Conceptualization, Writing-original draft, Visualization; Q.W.: Writing-review & editing; G.W.: Writing-review & editing; S.-T.Y.: Conceptualization, Writing-review & editing, Visualization, Supervision, Project administration, Funding acquisition.
Ethics Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The original contributions presented in the study are included in the article; further inquiries can be directed to the corresponding author.
Funding
This work was supported by the Advanced Research Projects Agency–Energy (ARPA-E) (Grant number: DE-AR0001512) and the US Department of Energy—Office of Energy Efficiency & Renewable Energy (DOE-EERE) (Grant number: EE0010300).
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
1.
Birgen C, Dürre P, Preisig HA, Wentzel A. Butanol production from lignocellulosic biomass: Revisiting fermentation performance indicators with exploratory data analysis.
Biotechnol. Biofuels 2019,
12, 167.
[Google Scholar]
2.
Niemistö J, Saavalainen P, Isomäki R, Kolli T, Huuhtanen M, Keiski RL. Biobutanol production from biomass. In Biofuel Technologies: Recent Developments; Gupta VK, Tuohy MG, Eds.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 443–470.
3.
Jones DT, Woods DR. Acetone-butanol fermentation revisited.
Microbiol. Rev. 1986,
50, 484–524.
[Google Scholar]
4.
Moon HG, Jang YS, Cho C, Lee J, Binkley R, Lee SY. One hundred years of clostridial butanol fermentation.
FEMS Microbiol. Lett. 2016,
363, fnw001.
[Google Scholar]
5.
Zhao J, Lu C, Chen CC, Yang ST. Biological production of butanol and higher alcohols. In Bioprocessing Technologies in Biorefinery for Sustainable Production of Fuels, Chemicals, and Polymers; Yang ST, El-Enshasy HA, Thongchul N, Eds.; John Wiley & Sons: New York, NY, USA, 2013; pp. 235–261.
6.
Veza I, Said MFM, Latiff ZA. Recent advances in butanol production by acetone-butanol-ethanol (ABE) fermentation.
Biomass Bioenergy 2021,
144, 105919.
[Google Scholar]
7.
Jiménez-Bonilla P, Wang S, Whitfield T, Blersch D, Wang Y, Gonzalez-de-Bashan L-E, et al. Tolerance in solventogenic Clostridia for enhanced butanol production: Genetic mechanisms and recent strain engineering advances.
Syn. Biol. Eng. 2024,
2, 10007.
[Google Scholar]
8.
Wang JF, Yang XR, Chen CC, Yang ST. Engineering clostridia for butanol production from biorenewable resources: from cells to process integration.
Curr. Opin. Chem. Eng. 2014,
6, 43–54.
[Google Scholar]
9.
Xue C, Zhao XQ, Liu CG, Chen LJ, Bai FW. Prospective and development of butanol as an advanced biofuel.
Biotechnol. Adv. 2013,
31, 1575–1584.
[Google Scholar]
10.
Cheng C, Bao T, Yang ST. Engineering
Clostridium for improved solvents production: Recent progress and perspective.
Appl. Microbiol. Biotechnol. 2019,
103, 5549–5566.
[Google Scholar]
11.
Bao T, Feng J, Jiang W, Fu H, Wang J, Yang ST. Recent advances in n-butanol and butyrate production using engineered
Clostridium tyrobutyricum.
World J. Microbiol. Biotechnol. 2020,
36, 138.
[Google Scholar]
12.
Abdelaal AS, Yazdani SS. Engineering
E. coli to synthesize butanol.
Biochem. Soc. Trans. 2022,
50, 867–876. doi:10.1042/BST20211009.
[Google Scholar]
13.
Xu M, Zhao J, Yu L, Yang ST. Comparative genomic analysis of
Clostridium acetobutylicum for understanding the mutations contributing to enhanced butanol tolerance and production.
J. Biotechnol. 2017,
263, 36–44.
[Google Scholar]
14.
Xu M, Zhao J, Yu L, Tang IC, Xue C, Yang ST. Engineering
Clostridium acetobutylicum with a histidine kinase knockout for enhanced n-butanol tolerance and production.
Appl. Microbiol. Biotechnol. 2015,
99, 1011–1022.
[Google Scholar]
15.
Du G, Zhu C, Xu M, Yang ST, Xue C. Energy-efficient butanol production by
Clostridium acetobutylicum with histidine kinase knockouts to improve strain tolerance and process robustness.
Green Chem. 2021,
23, 2155–2168.
[Google Scholar]
16.
Du G, Wu Y, Kang W, Xu Y, Li S, Xue C. Enhanced butanol production in
Clostridium acetobutylicum by manipulating metabolic pathway genes.
Process Biochem. 2022,
114, 134–138.
[Google Scholar]
17.
Nguyen NPT, Raynaud C, Meynial-Salles I, Soucaille P. Reviving the Weizmann process for commercial n-butanol production.
Nat. Commun. 2018,
9, 3682.
[Google Scholar]
18.
Yu M, Zhang Y, Tang IC, Yang ST. Metabolic engineering of
Clostridium tyrobutyricum for n-butanol production.
Metab. Eng. 2011,
13, 373–382.
[Google Scholar]
19.
Du Y, Jiang W, Yu M, Tang IC, Yang ST. Metabolic process engineering of
Clostridium tyrobutyricum Δack-adhE2 for enhanced n-butanol production from glucose: Effects of methyl viologen on NADH availability, flux distribution, and fermentation kinetics.
Biotechnol. Bioeng. 2015,
112, 705–715.
[Google Scholar]
20.
Huang J, Du Y, Bao T, Lin M, Wang J, Yang S-T. Production of n-butanol from cassava bagasse hydrolysate by engineered
Clostridium tyrobutyricum overexpressing adhE2: Kinetics and cost analysis.
Bioresour. Technol. 2019,
292, 121969.
[Google Scholar]
21.
Li J, Du Y, Bao T, Dong J, Lin M, Shim H, Yang ST. n-Butanol production from lignocellulosic biomass hydrolysates without detoxification by
Clostridium tyrobutyricum Δack-adhE2 in a fibrous-bed bioreactor.
Bioresour. Technol. 2019,
289, 121749.
[Google Scholar]
22.
Yu L, Zhao J, Xu M, Dong J, Varghese S, Yu M, et al. Metabolic engineering of
Clostridium tyrobutyricum for n-butanol production: effects of CoA transferase.
Appl. Microbiol. Biotechnol. 2015,
99, 4917–4930.
[Google Scholar]
23.
Zhang J, Yu L, Lin M, Yan Q, Yang ST. n-Butanol production from sucrose and sugarcane juice by engineered
Clostridium tyrobutyricum overexpressing sucrose catabolism genes and adhE2.
Bioresour. Technol. 2017,
233, 51–57.
[Google Scholar]
24.
Yu L, Xu M, Tang I-C, Yang S-T. Metabolic engineering of
Clostridium tyrobutyricum for n-butanol production from maltose and soluble starch by overexpressing α-glucosidase.
Appl. Microbiol. Biotechnol. 2015,
99, 6155–6165.
[Google Scholar]
25.
Yu L, Xu M, Tang IC, Yang ST. Metabolic engineering of
Clostridium tyrobutyricum for n-butanol production through co-utilization of glucose and xylose.
Biotechnol. Bioeng. 2015,
112, 2134–2141.
[Google Scholar]
26.
Zhang J, Zong W, Hong W, Zhang ZT, Wang Y. Exploiting endogenous CRISPR-Cas system for multiplex genome editing in
Clostridium tyrobutyricum and engineer the strain for high-level butanol production.
Metab. Eng. 2018,
47, 49–59.
[Google Scholar]
27.
Cao X, Chen Z, Liang L, Guo L, Jiang Z, Tang F, et al. Co-valorization of paper mill sludge and corn steep liquor for enhanced n-butanol production with
Clostridium tyrobutyricum Δcat1::adhE2.
Bioresour. Technol. 2020,
296, 122347.
[Google Scholar]
28.
Fu H, Hu J, Guo X, Feng J, Yang ST, Wang J. Butanol production from
Saccharina japonica hydrolysate by engineered
Clostridium tyrobutyricum: The effects of pretreatment method and heat shock protein overexpression,
Bioresource Technol. 2021,
335, 125290.
[Google Scholar]
29.
Atsumi S, Cann AF, Connor MR, Shen CR, Smith KM, Brynildsen MP, et al. Metabolic engineering of
Escherichia coli for 1-butanol production
Metab. Eng. 2008,
10, 305–311.
[Google Scholar]
30.
Shen CR, Lan EI, Dekishima Y, Baez A, Cho KM, Liao JC. Driving forces enable high-titer anaerobic 1-butanol synthesis in
Escherichia coli.
Appl. Environ. Microbiol. 2011,
77, 2905–2915.
[Google Scholar]
31.
Dong H, Zhao C, Zhang T, Zhu H, Lin Z, Tao W, et al. A systematically chromosomally engineered
Escherichia coli efficiently produces butanol.
Metab. Eng. 2017,
44, 284–292.
[Google Scholar]
32.
Jawed K, Abdelaal AS, Koffas MAG, Yazdani SS. Improved butanol production using FASII pathway in
E. coli.
ACS Synth. Biol. 2020,
9, 2390–2398.
[Google Scholar]
33.
Dellomonaco C, Clomburg J, Miller E, Gonzalez R. Engineered reversal of the β-oxidation cycle for the synthesis of fuels and chemicals.
Nature 2011,
476, 355–359. doi:10.1038/nature10333.
[Google Scholar]
34.
Girbal L, Soucaille P. Regulation of
Clostridium acetobutylicum metabolism as revealed by mixed- substrate steady-state continuous cultures: Role of NADH/NAD ratio and ATP pool.
J. Bacteriol. 1994,
176, 6433–6438.
[Google Scholar]
35.
Ballongue J, Amine J, Masion E, Petitdemange H, Gay R. Induction of acetoacetate decarboxylase in
Clostridium acetobutylicum.
FEMS Microbiol. Lett. 1985,
29, 273–277.
[Google Scholar]
36.
Fond O, Matta-Ammouri G, Petitdemange H, Engasser JM. The role of acids on the production of acetone and butanol by
Clostridium acetobutylicum.
Appl. Microbiol. Biotechnol. 1985,
22, 195–200.
[Google Scholar]
37.
Terracciano JS, Kashket ER. Intracellular conditions required for initiation of solvent production by
Clostridium acetobutylicum.
Appl. Environ. Microbiol. 1986,
52, 86–91.
[Google Scholar]
38.
Bahl H, Andersch W, Braun K, Gottschalk G. Effect of pH and butyrate concentration on the production of acetone and butanol by
Clostridium acetobutylicum grown in continuous culture.
Eur. J. Appl. Microbiol. Biotechnol. 1982,
14, 17–20.
[Google Scholar]
39.
Compere AL, Griffith WL. Evaluation of substrates for butanol production.
Dev. Ind. Microbiol. 1979,
20, 6510996.
[Google Scholar]
40.
Rogers P. Genetics and biochemistry of
Clostridium relevant to development of fermentation processes.
Adv. Appl. Microbiol. 1986,
31, 1–60.
[Google Scholar]
41.
Ounine K, Petitdemange H, Raval G, Gay R. Acetone-butanol production from pentoses by
Clostridium acetobutylicum.
Biotechnol. Lett. 1983,
5, 605–610.
[Google Scholar]
42.
Foulquier C, Rivière A, Heulot M, Dos Reis S, Perdu C, Girbal L, et al. Molecular characterization of the missing electron pathways for butanol synthesis in
Clostridium acetobutylicum.
Nat. Commun. 2022,
13, 4691.
[Google Scholar]
43.
Yoo M, Bestel-Corre G, Croux C, Riviere A, Meynial-Salles I, Soucaille P. A quantitative system-scale characterization of the metabolism of
Clostridium acetobutylicum.
MBio 2015,
6, 1–12.
[Google Scholar]
44.
Crown SB, Indurthi DC, Ahn WS, Choi J, Papoutsakis ET, Antoniewicz MR. Resolving the TCA cycle and pentose-phosphate pathway of
Clostridium acetobutylicum ATCC 824: Isotopomer analysis,
in vitro activities and expression analysis.
Biotechnol. J. 2011,
6, 300–305.
[Google Scholar]
45.
Mann MS, Lütke-Eversloh T. Thiolase engineering for enhanced butanol production in
Clostridium acetobutylicum.
Biotechnol. Bioeng. 2013,
110, 887–897.
[Google Scholar]
46.
Jang Y-S, Lee JYJJ, Lee JYJJ, Park JH, Im JA, Eom M, et al. Enhanced butanol production obtained by reinforcing the direct butanol-forming route in
Clostridium acetobutylicum.
MBio 2012,
3, 1–9.
[Google Scholar]
47.
Sillers R, Al-Hinai MA, Papoutsakis ET. Aldehyde-alcohol dehydrogenase and/or thiolase overexpression coupled with CoA transferase downregulation lead to higher alcohol titers and selectivity in
Clostridium acetobutylicum fermentations.
Biotechnol. Bioeng. 2009,
102, 38–49.
[Google Scholar]
48.
Du G, Che J, Wu Y, Wang Z, Jiang Z, Ji F, et al. Disruption of hydrogenase gene for enhancing butanol selectivity and production in
Clostridium acetobutylicum.
Biochem. Eng. J. 2021,
171, 108014.
[Google Scholar]
49.
Liu D, Yang Z, Wang P, Niu H, Zhuang W, Chen Y, et al. Towards acetone-uncoupled biofuels production in solventogenic
Clostridium through reducing power conservation.
Metab. Eng. 2018,
47, 102–112.
[Google Scholar]
50.
Liu T, Guo R, Wang X, Gu N, Wu N, Wu J, et al. Enhanced butanol production through intracellular NADH regeneration in CdSe-
C. acetobutylicumg semi-photosynthetic biohybrid system.
Bioresour. Technol. 2025,
418, 131939.
[Google Scholar]
51.
Zhou Z, Ding H, Shi C, Peng S, Zhu B, An X, et al. Enhanced butanol tolerance and production from puerariae slag hydrolysate by
Clostridium beijerinckii through metabolic engineering and process regulation strategies.
Bioresour. Technol. 2025,
419, 132035.
[Google Scholar]
52.
Chang WL, Hou W, Xu M, Yang ST. High-rate continuous n-butanol production by
Clostridium acetobutylicum from glucose and butyric acid in a single-pass fibrous-bed bioreactor.
Biotechnol. Bioeng. 2022,
119, 3474–3486.
[Google Scholar]
53.
Ehsaan M, Yoo M, Kuit W, Foulquier C, Soucaille P, Minton NP. Chromosomal integration of the pSOL1 megaplasmid of
Clostridium acetobutylicum for continuous and stable advanced biofuels production.
Nat. Microbiol. 2024,
9, 1655–1660.
[Google Scholar]
54.
Yang Y, Nie X, Jiang Y, Yang C, Gu Y, Jiang W. Metabolic regulation in solventogenic clostridia: Regulators, mechanisms and engineering.
Biotechnol. Adv. 2018,
36, 905–914.
[Google Scholar]
55.
Al-Hinai MA, Jones SW, Papoutsakis ET. The
Clostridium sporulation programs: diversity and preservation of endospore differentiation.
Microbiol. Mol. Biol. Rev. 2015,
79, 19–37.
[Google Scholar]
56.
Diallo M, Kengen SWM, Lopez-Contreras AM. Sporulation in solventogenic and acetogenic clostridia.
Appl. Microbiol. Biotechnol. 2021,
105, 3533–3557.
[Google Scholar]
57.
Alsaker KV, Spitzer TR, Papoutsakis ET. Transcriptional of spo0A overexpression in
Clostridium acetobutylicum and its effect on the cell’s response to butanol stress.
J .Bacteriol. 2004,
186, 1959–1971.
[Google Scholar]
58.
Steiner E, Dago AE, Young DI, Heap JT, Minton NP, Hoch JA, et al. Multiple orphan histidine kinases interact directly with Spo0A to control the initiation of endospore formation in
Clostridium acetobutylicum.
Mol. Microbiol. 2011,
80, 641–654.
[Google Scholar]
59.
Zhu C, Du G, Zhang J, Xue C. A high-efficient strategy for combinatorial engineering paralogous gene family: A case study on histidine kinases in
Clostridium.
Biotechnol. Bioeng. 2021,
118, 2770–2780.
[Google Scholar]
60.
Du G, Zhu C, Wu Y, Kang W, Xu M, Yang ST, et al. Effects of orphan histidine kinases on clostridial sporulation progression and metabolism.
Biotechnol. Bioeng. 2022,
119, 226–235.
[Google Scholar]
61.
Zhu C, Wang Z, Zhou X, Wu Y, Kang W, Wu R, et al. Elucidating the biosynthesis and function of an autoinducing peptide in
Clostridium acetobutylicum.
Angewantge 2025, e202500904. doi:10.1002/anie.202500904.
[Google Scholar]
62.
Lee J, Jang YS, Han MJ, Kim JY, Lee SY. Deciphering
Clostridium tyrobutyricum metabolism based on the whole-genome sequence and proteome analyses.
MBio 2016,
7, e00743-16.
[Google Scholar]
63.
Dwidar M, Park J-Y, Mitchell RJ, Sang B-I. The future of butyric acid in industry.
Sci. World J. 2012,
2012, 471417.
[Google Scholar]
64.
Jiang L, Fu H, Yang HK, Xu W, Wang J, Yang S-T. Butyric acid: Applications and recent advances in its bioproduction.
Biotechnol. Adv. 2018,
36, 2101–2117.
[Google Scholar]
65.
Yu M, Du Y, Jiang W, Chang WL, Yang ST, Tang IC. Effects of different replicons in conjugative plasmids on transformation efficiency, plasmid stability, gene expression and n-butanol biosynthesis in
Clostridium tyrobutyricum.
Appl. Microbiol. Biotechnol. 2012,
93, 881–889.
[Google Scholar]
66.
Ferreira S, Pereira R, Wahl SA, Rocha I. Metabolic engineering strategies for butanol production in
Escherichia coli.
Biotechnol. Bioeng. 2020,
117, 2571–2587.
[Google Scholar]
67.
Koppolu V, Vasigala VKR. Role of
Escherichia coli in biofuel production.
Microbiol. Insights 2016,
9, MBI.S10878.
[Google Scholar]
68.
Dong H, Zhao C, Zhang T, Lin Z, Li Y, Zhang Y. Engineering
Escherichia coli cell factories for n-butanol production.
Adv. Biochem. Eng./Biotechnol. 2016,
1155, 141–163.
[Google Scholar]
69.
Ohta K, Beall DS, Mejia JP, Shanmugam KT, Ingram LO. Genetic improvement of
Escherichia coli for ethanol production: Chromosomal integration of
Zymomonas mobilis genes encoding pyruvate decarboxylase and alcohol dehydrogenase II.
Appl. Environ. Microbiol. 1991,
57, 893–900.
[Google Scholar]
70.
Pásztor A, Kallio P, Malatinszky D, Akhtar MK, Jones PR. A synthetic O
2-tolerant butanol pathway exploiting native fatty acid biosynthesis in
Escherichia coli.
Biotechnol. Bioeng. 2015,
112, 120–128.
[Google Scholar]
71.
Gross GG. Evidence for enzyme-substrate intermediates in the aryl-aldehyde: NADP oxidoreductase catalysed reduction of salicylate.
FEBS Lett. 1969,
5, 177–179.
[Google Scholar]
72.
Winkler M, Ling JG. Biocatalytic carboxylate reduction—Recent advances and new enzymes.
ChemCatChem 2022,
14, e202200441.
[Google Scholar]
73.
Gross GG, Bolkart KH, Zenk MH. Reduction of cinnamic acid to cinnamaldehyde and alcohol.
Biochem. Biophys. Res. Commun. 1968,
32, 173–178.
[Google Scholar]
74.
Duan Y, Yao P, Chen X, Liu X, Zhang R, Feng J, et al. Exploring the synthetic applicability of a new carboxylic acid reductase from
Segniliparus rotundus DSM 44985.
J. Mol. Catal. B Enzym. 2015,
115, 1–7.
[Google Scholar]
75.
Venkitasubramanian P, Daniels L, Rosazza J. Biocatalytic reduction of carboxylic acids: Mechanism and applications. In Biocatalysis in the Pharmaceutical and Biotechnology Industries; Patel RN, Ed.; CRC Press: Boca Raton, FL, USA, 2006; pp. 425–440.
76.
He A, Li T, Daniels L, Fotheringham I, Rosazza JPN.
Nocardia sp. carboxylic acid reductase: Cloning, expression, and characterization of a new aldehyde oxidoreductase family.
Appl. Environ. Microbiol. 2004,
70, 1874–1881.
[Google Scholar]
77.
Moore CD. Metabolic and Protein Engineering of Butanol-Producing C. tyrobutyricum. PhD thesis, The Ohio State University, Columbus, OH, USA, 2024.
78.
Wilson KR, Prophet AM, Rovelli G, Willis MD, Rapf RJ, Jacobs MI. A kinetic description of how interfaces accelerate reactions in micro-compartments.
Chem. Sci. 2020,
11, 8533–8545.
[Google Scholar]
79.
Wang L, Sun Y, Diao S, Jiang S, Wang H, Wei D. Rational hinge engineering of carboxylic acid reductase from
Mycobacterium smegmatis enhances its catalytic efficiency in biocatalysis.
Biotechnol. J. 2022,
17, 2100441.
[Google Scholar]
80.
Girbal L, Soucaille P. Regulation of solvent production in
Clostridium acetobutylicum.
Trends Biotechnol. 1998,
16, 11–16.
[Google Scholar]
81.
Vasconcelos I, Girbal L, Soucaille P. Regulation of carbon and electron flow in
Clostridium acetobutylicum grown in chemostat culture at neutral pH on mixtures of glucose and glycerol.
J. Bacteriol. 1994,
176, 1443–1450.
[Google Scholar]
82.
Sharma VK, Hutchison JM, Allgeier AM. Redox biocatalysis: Quantitative comparisons of nicotinamide cofactor regeneration methods.
ChemSusChem 2022,
15, e202200888.
[Google Scholar]
83.
Yoo YJ, Kim YH, Feng Y, Yagonia CFJ. Fundamentals of Enzyme Engineering; Springer: Berlin, Germany, 2017; pp. 1–209.
84.
Findrik Z, Šimunović I, Vasić-Rački D. Coenzyme regeneration catalyzed by NADH oxidase from
Lactobacillus brevis in the reaction of l-amino acid oxidation.
Biochem. Eng. J. 2008,
39, 319–327.
[Google Scholar]
85.
Bai Y, Yang ST. Biotransformation of R-2-hydroxy-4-phenylbutyric acid by D-lactate dehydrogenase and
Candida boidinii cells containing formate dehydrogenase coimmobilized in a fibrous bed bioreactor,
Biotechnol. Bioeng. 2005,
92, 137–146.
[Google Scholar]
86.
Khusnutdinova AN, Flick R, Popovic A, Brown G, Tchigvintsev A, Nocek B, et al. Exploring bacterial carboxylate reductases for the reduction of bifunctional carboxylic acids.
Biotechnol. J. 2017,
12, 1600751.
[Google Scholar]
87.
Zhang DP, Jing XR, Wu LJ, Fan AW, Nie Y, Xu Y. Highly selective synthesis of d-amino acids via stereoinversion of corresponding counterpart by an
in vivo cascade cell factory.
Microb. Cell Fact. 2021,
20, 11.
[Google Scholar]
88.
Abdel-Hady GN, Ikeda T, Ishida T, Funabashi H, Kuroda A, Hirota R. Engineering cofactor specificity of a thermostable phosphite dehydrogenase for a highly efficient and robust NADPH regeneration system.
Front. Bioeng. Biotechnol. 2021,
9, 647176.
[Google Scholar]
89.
Guo X, Wang X, Liu Y, Li Q, Wang J, Liu W, et al. Structure-guided design of formate dehydrogenase for regeneration of a non-natural redox cofactor.
Chem.—A Eur. J. 2020,
26, 16611–16615.
[Google Scholar]
90.
Black WB, Zhang L, Mak WS, Maxel S, Cui Y, King E, et al. Engineering a nicotinamide mononucleotide redox cofactor system for biocatalysis.
Nat. Chem. Biol. 2020,
16, 87–94.
[Google Scholar]
91.
Tavanti M, Hosford J, Lloyd RC, Brown MJB. ATP regeneration by a single polyphosphate kinase powers multigram-scale aldehyde synthesis
in vitro.
Green Chem. 2021,
23, 828–837.
[Google Scholar]
92.
Maier A, Mguni LM, Ngo ACR, Tischler D. Formate dehydrogenase: Recent developments for NADH and NADPH recycling in biocatalysis.
ChemCatChem 2024,
16, e202401021.
[Google Scholar]
93.
Lu Y, Zhang C, Zhao H, Xing XH. Improvement of hydrogen productivity by introduction of NADH regeneration pathway in
Clostridium paraputrificum.
Appl. Biochem. Biotechnol. 2012,
167, 732–742.
[Google Scholar]
94.
Han S, Gao XY, Ying HJ, Zhou CC. NADH gene manipulation for advancing bioelectricity in
Clostridium ljungdahlii microbial fuel cells.
Green Chem. 2016,
18, 2473–2478.
[Google Scholar]
95.
Ma C, Ou J, Xu N, Fierst JL, Yang ST, Liu X. Rebalancing redox to improve biobutanol production by
Clostridium tyrobutyricum.
Bioengineering 2015,
3, 2–15.
[Google Scholar]
96.
Mitamura T, Urabe I, Okada H. Enzymatic properties of isozymes and variants of glucose dehydrogenase from
Bacillus megaterium.
Eur. J. Biochem. 1989,
186, 389–393.
[Google Scholar]
97.
Hanson RL, Schwinden MD, Banerjee A, Brzozowski DB, Chen B-C, Patel BP, et al. Enzymatic synthesis of l-6-hydroxynorleucine.
Bioorg. Med. Chem. 1999,
7, 2247–2252.
[Google Scholar]
98.
Lin SS, Miyawaki O, Nakamura K. Continuous production of L-carnitine with NADH regeneration by a nanofiltration membrane reactor with coimmobilized L-carnitine dehydrogenase and glucose dehydrogenase.
J. Biosci. Bioeng. 1999,
87, 361–364.
[Google Scholar]
99.
Costas AM, White AK, Metcalf WW. Purification and characterization of a novel phosphorus-oxidizing enzyme from
Pseudomonas stutzeri WM88.
J. Biol. Chem. 2001,
276, 17429–17436.
[Google Scholar]
100.
White AK, Metcalf WW. Isolation and biochemical characterization of hypophosphite/2-oxoglutarate dioxygenase. A novel phosphorus-oxidizing enzyme from
Psuedomonas stutzeri WM88.
J. Biol. Chem. 2002,
277, 38262–38271.
[Google Scholar]
101.
Relyea HA, Van Der Donk WA. Mechanism and applications of phosphite dehydrogenase.
Bioorg. Chem. 2005,
33, 171–189.
[Google Scholar]
102.
White AK, Metcalf WW. The htx and ptx operons of
Pseudomonas stutzeri WM88 are new members of the Pho regulon.
J. Bacteriol. 2004,
186, 5876–5882.
[Google Scholar]
103.
Fogle EJ, van der Donk WA. Pre-steady-state studies of phosphite dehydrogenase demonstrate that hydride transfer is fully rate limiting.
Biochemistry 2007,
46, 13101–13108.
[Google Scholar]
104.
Woodyer R, Van der Donk WA, Zhao H. Relaxing the nicotinamide cofactor specificity of phosphite dehydrogenase by rational design.
Biochemistry 2003,
42, 11604–11614.
[Google Scholar]
105.
Girbal L, Croux C, Vasconcelos I, Soucaille P. Regulation of metabolic shifts in
Clostridium acetobutylicum ATCC 824.
FEMS Microbiol. Rev. 1995,
17, 287–297.
[Google Scholar]
106.
Dürre P, Fischer RJ, Kuhn A, Lorenz K, Schreiber W, Stürzenhofecker B, et al. Solventogenic enzymes of
Clostridium acetobutylicum: catalytic properties, genetic organization, and transcriptional regulation.
FEMS Microbiol. Rev. 1995,
17, 251–262.
[Google Scholar]
107.
Hempel J, Harper K, Lindahl R. Inducible (class 3) aldehyde dehydrogenase from rat hepatocellular carcinoma and 2,3,7,8-tetrachlorodibenzo-p-dioxin-treated liver: distant relationship to the class 1 and 2 enzymes from mammalian liver cytosol/mitochondria.
Biochemistry 1989,
28, 1160–1167.
[Google Scholar]
108.
Yoo M, Croux C, Salles IM, Soucaille P. Elucidation of the roles of adhE1 and adhE2 in the primary metabolism of
Clostridium acetobutylicum by combining in-frame gene deletion and a quantitative system-scale approach.
Biotechnol. Biofuels 2016,
9, 92.
[Google Scholar]
109.
Extance J, Crennell SJ, Eley K, Cripps R, Hough DW, Danson MJ. Structure of a bifunctional alcohol dehydrogenase involved in bioethanol generation in
Geobacillus thermoglucosidasius.
Acta Crystallogr. Sect. D Biol. Crystallogr. 2013,
69, 2104–2115.
[Google Scholar]
110.
Rellos P, Ma J, Scopes RK. Alteration of substrate specificity of
Zymomonas mobilis alcohol dehydrogenase-2 using
in vitro random mutagenesis.
Protein Expr. Purif. 1997,
9, 83–90.
[Google Scholar]
111.
Cho C, Hong S, Moon HG, Jang YS, Kim D, Lee SY. Engineering clostridial aldehyde/alcohol dehydrogenase for selective butanol production.
MBio 2019,
10, 1–12.
[Google Scholar]
112.
Zheng T, Olson DG, Tian L, Bomble YJ, Himmel ME, Lo J, et al. Cofactor specificity of the bifunctional alcohol and aldehyde dehydrogenase (AdhE) in wild-type and mutant
Clostridium thermocellum and
thermoanaerobacterium saccharolyticum.
J. Bacteriol. 2015,
197, 2610–2619.
[Google Scholar]
113.
Solanki K, Abdallah W, Banta S. Engineering the cofactor specificity of an alcohol dehydrogenase via single mutations or insertions distal to the 2′-phosphate group of NADP(H).
Protein Eng. Des. Sel. 2017,
30, 373–380.
[Google Scholar]
114.
Jiang Y, Li X, Liu B, Tong F, Qu G, Sun Z. Engineering the hydrogen transfer pathway of an alcohol dehydrogenase to increase activity by rational enzyme design.
Mol. Catal. 2022,
530, 112603.
[Google Scholar]
115.
Schmitz S, Hupfeld E, Sterner R, Merkl R, Lo P. Rosetta:MSF: A modular framework for multi-state computational protein design.
PLoS Comput. Biol. 2017,
13, e1005600.
[Google Scholar]
116.
Guarneri A, van Berkel WJ, Paul CE. Alternative coenzymes for biocatalysis.
Curr. Opin. Biotechnol. 2019,
60, 63–71.
[Google Scholar]
117.
Zachos I, Döring M, Tafertshofer G, Simon RC, Sieber V. carba Nicotinamide adenine dinucleotide phosphate: Robust cofactor for redox biocatalysis.
Angew. Chem. Int. Ed. Engl. 2021,
60, 14701–14706.
[Google Scholar]
118.
Weusthuis RA, Folch PL, Pozo-Rodríguez A, Paul CE. Applying non-canonical redox cofactors in fermentation processes.
iScience 2020,
23, 101471.
[Google Scholar]
119.
Aspacio D, Zhang Y, Cui Y, Luu E, King E, Black WB, et al. Shifting redox reaction equilibria on demand using an orthogonal redox cofactor.
Nat. Chem. Biol. 2024,
20, 1535–1546.
[Google Scholar]
120.
Resnick SM, Zehnder AJB.
In vitro ATP regeneration from polyphosphate and AMP by polyphosphate:AMP phosphotransferase and adenylate kinase from
Acinetobacter johnsonii 210A.
Appl. Environ. Microbiol. 2000,
66, 2045–2051.
[Google Scholar]
121.
Neville N, Roberge N, Jia Z. Polyphosphate kinase 2 (PPK2) enzymes: Structure, function, and r in bacterial physiology and virulence.
Int. J. Mol. Sci. 2022,
23, 670.
[Google Scholar]
122.
Köpke M, Held C, Hujer S, Liesegang H, Wiezer A, Wollherr A, et al.
Clostridium ljungdahlii represents a microbial production platform based on syngas.
Proc. Natl. Acad. Sci. USA 2010,
107, 13087–13092.
[Google Scholar]
123.
Liew F, Henstra AM, Köpke M, Winzer K, Simpson SD, Minton NP. Metabolic engineering of
Clostridium autoethanogenum for selective alcohol production.
Metab Eng. 2017,
40, 104–114.
[Google Scholar]
124.
Mock J, Zheng Y, Mueller AP, Ly S, Tran L, Segovia S, et al. Energy conservation associated with ethanol formation from H
2 and CO
2 in
Clostridium autoethanogenum involving electron bifurcation.
J. Bacteriol. 2015,
197, 2965–2980.
[Google Scholar]
125.
Xu H, Liang C, Chen X, Xu J, Yu Q, Zhang Y, et al. Impact of exogenous acetate on ethanol formation and gene transcription for key enzymes in
Clostridium autoethanogenum grown on CO.
Biochem. Eng. J. 2020,
155, 107470.
[Google Scholar]
126.
Zhu HF, Liu ZY, Zhou X, Yi JH, Lun ZM, Wang SN, et al. Energy conservation and carbon flux distribution during fermentation of CO or H
2/CO
2 by
Clostridium ljungdahlii.
Front. Microbiol. 2020,
11, 416.
[Google Scholar]
127.
Bao T, Cheng C, Xin X, Wang J, Wang M, Yang ST. Deciphering mixotrophic
Clostridium formicoaceticum metabolism and energy conservation: Genomic analysis and experimental studies.
Genomics 2019,
111, 1687–1694.
[Google Scholar]
128.
Cheng C, Li W, Lin M, Yang ST. Metabolic engineering of
Clostridium carboxidivorans for enhanced ethanol and butanol production from syngas and glucose,
Bioresour. Technol. 2019,
284, 415–423.
[Google Scholar]
129.
Fraisse L, Simon H. Observations on the reduction of non-activated carboxylates by
Clostridium formicoaceticum with carbon monoxide or formate and the influence of various viologens.
Arch. Microbiol. 1988,
150, 381–386.
[Google Scholar]
130.
Ragsdale SW, Pierce E. Acetogenesis and the Wood–Ljungdahl pathway of CO
2 fixation.
Biochim. Biophys. Acta (BBA)—Proteins Proteom. 2008,
1784, 1873–1898.
[Google Scholar]
131.
Jones SW, Fast AG, Carlson ED, Wiedel CA, Au J, Antoniewicz MR, et al. CO
2 fixation by anaerobic non-photosynthetic mixotrophy for improved carbon conversion.
Nat. Comm. 2016,
7, 12800.
[Google Scholar]
132.
Tang IC, Yang ST, Okos MR. Acetic acid production from whey lactose by the co-culture of
Streptococcus lactis and
Clostridium formicoaceticum.
Appl. Microbiol. Biotechnol. 1988,
28, 138–143.
[Google Scholar]
133.
Yang ST, Tang IC, Okos MR. Kinetics of homoacetic fermentation of lactate by
Clostridium formicoaceticum.
Appl. Environ. Microbiol. 1987,
53, 823–827.
[Google Scholar]
134.
Du D, Lan R, Humphreys J, Tao S. Progress in inorganic cathode catalysts for electrochemical conversion of carbon dioxide into formate or formic acid.
J. Appl. Electrochem. 2017,
47, 661–678.
[Google Scholar]
135.
Fu H, Lin M, Tang IC, Wang J, Yang ST. Effects of benzyl viologen on increasing NADH availability, acetate assimilation and butyric acid production by
Clostridium tyrobutyricum.
Biotechnol. Bioeng. 2021,
118, 770–783.
[Google Scholar]