1. Introduction
Metabolism has been proposed to play an important role in inflammation and immunity [
1,
2]. Intracellular metabolism has a profound impact on T-cell function, which supports cell survival and activation by meeting the bioenergetic and biosynthetic [
3], such as the memory T cells and iTreg cells rely on lipid oxidation as a major energy source and effector T cells sustain high glycolytic activity and glutaminolytic activity [
4]. Naive T cells are metabolically quiescent, with low rates of oxygen consumption and glucose consumption. However, concomitant with T cell activation is the engagement of aerobic glycolysis and elevated oxidative phosphorylation [
5,
6]. Therefore, any factors affecting the metabolism of T cells will have an impact on the function of T cells.
Cylindromatosis (CYLD) is a deubiquitinating enzyme widely distributed in the body and is found in low levels in a large part of somatic cells, with immune cells expressing more of it [
7,
8]. Additionally, CYLD is a key regulator of diverse cellular processes such as immune responses, inflammation and proliferation [
9,
10,
11]. It can deubiquitize signaling molecules such as TRAFs, NF-κB essential modulator (NEMO), B cell lymphoma 3 protein (BCL3) and P53, and has been reported as a key negative modulator for NF-κB signaling [
12,
13,
14]. Meanwhile, it has been found that CYLD plays a crucial role in T cell development and tumor cell proliferation [
9,
15,
16].
Autophagy is a process related to breaking down and reusing cytoplasm components and is an essential cellular response in the fight against intracellular pathogens [
15,
17]. Microtubule-associated protein light chain3 (LC3-I) is conjugated to phosphatidyl ethanolamine to form LC3-II, which is a widely used autophagy marker and complete autophagosome marker. P62 is a ubiquitin-binding protein known as an autophagy substrate and is efficiently degraded κin the process of autophagy [
18]. Studies have shown that CYLD interacts directly with P62 and directly inactivates histone deacetylase(HDAC6), thereby controlling autophagy [
19]. In addition to ubiquitin-regulated P62, beclin1 can interact through CYLD-related TRAF6. In this way, CYLD can affect autophagy pathways [
20].
At the same time, many studies have shown that autophagy can be regulated by the AMPKα/mTOR/ULK1 signaling pathway [
21]. Impaired autophagy also prevents the degradation of lipid droplets leading to lipid deposition. AMPKα is the center of energy metabolism [
22], and mitochondria are the energy center of cells. Studies have shown that AMPKα can enhance mitochondrial energy metabolism and improve mitochondrial function by regulating the number of mitochondria and increasing enzyme activity in mitochondria, while mitochondrial dysfunction leads to metabolic disorders. While, whether CYLD regulates the pathway of autophagy and affects cell metabolism through the AMPKα signaling pathway is still unknown.
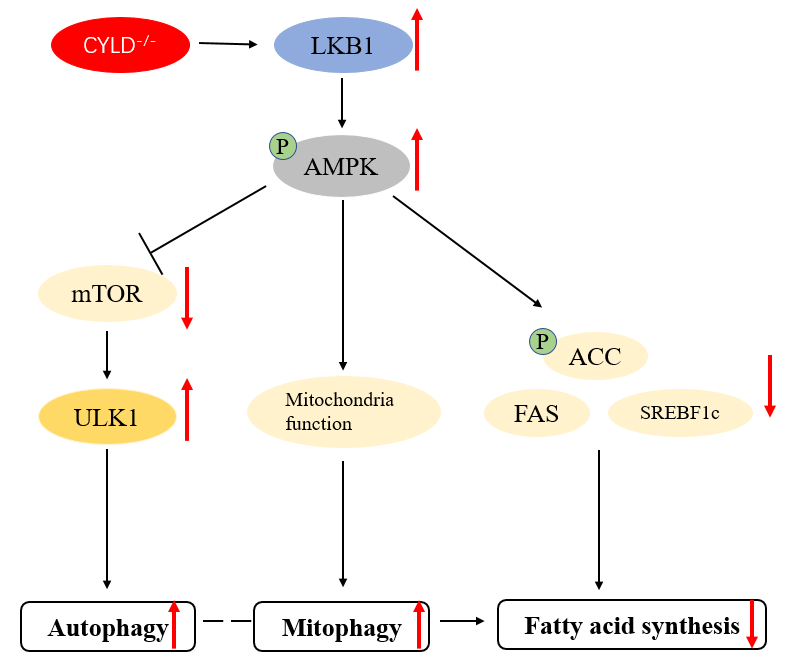
Herein, we have explored whether CYLD affects autophagy through AMPKα/mTOR/ULK1 signaling pathway and whether CYLD deletion affects mitochondrial autophagy and cellular metabolism via the AMPKα signaling pathway in T cells. First, we used the Jurkat cell model to knock out the CYLD gene and detected the potential correlation between autophagy and CYLD by detecting autophagy-related markers. Second, we found that CYLD regulates autophagy via the AMPKα/mTOR/ULK1 signaling pathway. Third, we found that CYLD deletion affects mitochondrial function and improves cell metabolism through AMPKα signaling pathway. Our study suggests CYLD deletion induces autophagy, affects mitochondrial function and improves lipid metabolism through AMPKα signaling pathway.
2. Materials and Methods
2.1. Cell Culture
Jurkat cells were purchased from ATCC (Rockefelle, MD, USA). Cylindromatosis (CYLD) knockout in Jurkat cells was generated by CRISPR/Cas9 gene editing system. All cells were cultured in RPMI-1640 medium (TBD) supplemented with 10% FBS (Gibico, NY, USA), 18 μg/mL penicillin G potassium (TBD), and 50 μg/mL streptomycin sulfate (Meiji Seika, Tokyo, Japan) at 37 °C in an atmosphere of 5% CO
2.
2.2. CRISPR-Cas9
Design sgRNA: Design sgRNA (single directional RNA) for the target gene to ensure that it does not have predicted off-target binding sites. Then constructing sgRNA-Cas9 vector: cloning the selected sgRNA into the corresponding expression vector. Constructing stable transgenic strains of sgRNA-Cas9: Using a lentiviral packaging system to construct stable cell lines expressing sgRNA-Cas9, and conducting puromycin screening to isolate cell clones. Screening and validation of cell clones: Perform quantitative PCR validation on the selected cell clones to confirm the occurrence of gene editing. Positive clone sequencing: Sequencing of clones confirmed positive by qPCR, selecting knockout homozygous cell lines for subsequent research. Cell monoclonal sequencing: further screening of cell monoclonal antibodies and sequencing analysis to ensure the accuracy of gene knockout.
2.3.Cell Viability Assay
Cell viability was determined using a CCK-8 assay kit (Kermay, Mc0301, Suqian, China) according to the manufacturer’s instructions. Briefly, 5 × 10
4 Jurkat cells in 100 μL culture media were plated to a 96-well plate in suspension and cultured for 0, 24, 48, and 72 h in an incubator. Then, the samples were incubated with CCK-8 solution (10 μL) for 4 h at 37 °C; the absorbance in each well was quantified at 450 nm using an automated enzyme-linkedimmunosorbent assay reader.
2.4. Western Blotting
The cells were lysed for 30 min in radio immunoprecipitation assay lysis buffer (Ripa buffer) (Beyotime, Shanghai, China) containing protease inhibitors on the ice and then centrifuged at 12,000×
g for 15 min at 4 ℃. Proteins were separated by 8–15% sodium dodecyl sulfate polyacrylamide gel electrophoresis and blotted onto a polyvinylidene difluoride membrane (PVDF) (Millipore, Burlington, MA, USA) following blocking with 5% nonfat dry milk in tris buffered saline (TBS) with 0.05% Tween 20 (TBST) at room temperature for 2 h and then incubated with the primary antibodies (dilution, 1:2000) overnight at 4 °C. Then the PVDF membranes were washed three times for 6 min in TBST on the shaker. After washing, the membranes were incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies (dilution, 1:3000) at room temperature for 1 h and the PVDF membranes were then washed three times for 6 min in TBST. Mitochondrial proteins were extracted according to the manufacturer’s protocol (Beyotime). Following antibodies (Abs) were used in this study: CYLD (#CST-8462), LC3 (#CST 3868), Beclin 1 (#CST 3495), AMPKα (#Proteintech-10929-2-AP), phosphor-AMPKα (P-AMPKα; #CST-2535), total mammalian target of rapamycin (mTOR; #CST 2983), phospho mTOR (P-mTOR; #CST 5536), Unc 51 like autophagy activating kinase 1 (ULK1; #Proteintech-66536), phospho ULK1 (P-ULK1; #CST 5869), PGC-1α (#CST 2178), Phospho-Acetyl-CoA Carboxylase (Ser79) (P-ACC; #CST D7D11), Acetyl-CoA Carboxylase (ACC; #CST C83B10), Fatty Acid Synthase (FAS; #CST-C20G5), GAPDH (#ab-9485), and anti rabbit IgG (#CST 2385) (Cell Signaling Technology, MA, USA).
2.5. RNA Isolation and Quantitative Real-Time PCR
Total RNA was extracted from cells with Trizol (Invitrogen, Carlsbad, CA, USA) reagent. The RNA concentration was measured using a spectrophotometer (Thermo Scientific, Waltham, MA, USA). First-strand complementary DNA (cDNA) was obtained using the PrimeaScript™ II First-Strand cDNA synthesis kit (TransGen Biotech, Beijing, China), according to the manufacturer’s instructions. Subsequently, SYBR green-based quantitative real-time PCR (qPCR) was performed using a power SYBR green master mix (TOYOBO) and 7500 Fast Real-Time PCR system (Thermofisher, Waltham, MA, USA). The relative mRNA level was expressed as fold change relative to untreated controls after normalization to the expression of GAPDH by the 2
−ΔΔCT method. The sequences of the primers used for PCR are listed in .
Table 1. Primers Used for Real-Time PCR.
Target Genes |
Primer Pairs 5′–3′ |
Forward |
Reverse |
| CYLD |
TCAGGCTTATGGAGCCAAGAA |
ACTTCCCTTCGGTACTTTAAGGA |
| Beclin1 |
CCATGCAGGTGAGCTTCGT |
GAATCTGCGAGAGACACCATC |
| P62 |
GCACCCCAATGTGATCTGC |
CGCTACACAAGTCGTAGTCTGG |
| ULK1 |
GGCAAGTTCGAGTTCTCCCG |
CGACCTCCAAATCGTGCTTCT |
| FAS |
TCTGGTTCTTACGTCTGTTGC |
CTGTGCAGTCCCTAGCTTTCC |
| ACC |
CATGCGGTCTATCCGTAGGTG |
GTGTGACCATGACAACGAATCT |
| SREBF1c |
CGGAACCATCTTGGCAACAGT |
CGCTTCTCAATGGCGTTGT |
| ADRP |
ATGGCATCCGTTGCAGTTGAT |
GGACATGAGGTCATACGTGGAG |
| HSL |
TCAGTGTCTAGGTCAGACTGG |
AGGCTTCTGTTGGGTATTGGA |
| CPT1A |
TCCAGTTGGCTTATCGTGGTG |
TCCAGAGTCCGATTGATTTTTGC |
| IL-4 |
CCAACTGCTTCCCCCTCTG |
TCTGTTACGGTCAACTCGGTG |
| IL-10 |
GACTTTAAGGGTTACCTGGGTTG |
TCACATGCGCCTTGATGTCTG |
| IL-13 |
CCTCATGGCGCTTTTGTTGAC |
TCTGGTTCTGGGTGATGTTGA |
| TNF-α |
GAGGCCAAGCCCTGGTATG |
CGGGCCGATTGATCTCAGC |
| IFN-γ |
TCGGTAACTGACTTGAATGTCCA |
TCGCTTCCCTGTTTTAGCTGC |
| IL-5 |
TGGAGCTGCCTACGTGTATG |
TTCGATGAGTAGAAAGCAGTGC |
| GAPDH |
CTGGGCTACACTGAGCACC |
AAGTGGTCGTTGAGGGCAATG |
2.6. Measurement of Intracellular ROS Level
According to the manufacturer’s instructions, prepare a 500× stock solution of the ROS Assay Stain by adding 40 μL of DMSO to the vial of ROS Assay Stain Concentrate and mixing well. The 500× ROS Assay Stain stock solution should be used at 1× to label cells. It may be added directly to cells in culture media at a final concentration of 1× by adding 2 μL of the 500× ROS Assay Stain stock solution for every 1 mL of cells; mix well. Alternatively, the 500× ROS Assay Stain stock solution may be diluted to 1× using the ROS Assay Buffer. For each sample, you will need 100 μL of ROS Assay Stain Solution. For example, for 10 experimental samples, you will need 1 mL of 1× ROS Assay Stain. Therefore, add 2 μL of 500× ROS Assay Stain to 1 mL of ROS Assay Buffer. Then use 100 μL of 1× ROS Assay Stain to resuspend the cells. Incubate for 60 min in a 37 °C incubator with 5% CO
2. Treat cells with the desired reagents to induce the production of ROS. Analyze on a flow cytometer at the desired time points.
2.7. Analysis of Mitochondrial Respiration
Mitochondrial bioenergetics was evaluated using the Seahorse XFp Extracellular Flux Analyzer (Seahorse Biosciences, XF24, San Francisco, CA, USA). A classical Mitostress test was performed according to the following procedure. Briefly, we put approximately the same number of cells in each well. At 1 h before the measurements, the growth medium was replaced with the experimental medium, and then cells were treated consecutively with oligomycin (an ATP synthase inhibitor, 2 µg/mL), FCCP (an uncoupling agent that collapses the proton gradient and disrupts the mitochondrial membrane potential, 2 μM), and antimycin A and rotenone (a complex III inhibitor and a complex I inhibitor, respectively, 4 μM). Basal respiration was measured in the unbuffered medium; ATP regeneration-driven O
2 consumption rate (OCR) was calculated by subtracting OCR in the presence of oligomycin; maximal respiration, an index of spare respiratory capacity, was quantified in the presence of FCCP; and respiratory reserve capacity was calculated by subtracting basal OCR from maximal OCR.
2.8. Immunofluorescence
The cells were harvested into EP tubes, fixed with 4% paraformaldehyde in PBS for 20 min and then washed 3 times with PBS. Then cells were resuspended with 5% BSA for 30 min, and incubated with primary antibody (1:200) for 2 h. After washing 3 times with PBS, the second antibody (1:100) was incubated for 1 h and stained with DAPI for 10 min. After washing with PBS, the cells were applied to the adhering slides and the coverslips were observed when slightly moistened.
2.9. Confocal Microscopy
Both immunostained and live cells were observed with a confocal laser scanning microscope (Zeiss LSM510, Zeiss GmbH, Germany). Live cells were imaged in a temperature-controlled chamber (37 ℃) at 5% CO
2.
2.10. Statistical Analysis
Each experiment was performed at least three times, and the results were expressed as the mean ± standard deviation (SD). Statistical analysis was performed with the student
t test, and
p < 0.05 was considered significant.
3. Results
3.1. CYLD Deletion Induces Autophagy in Jurkat Cells
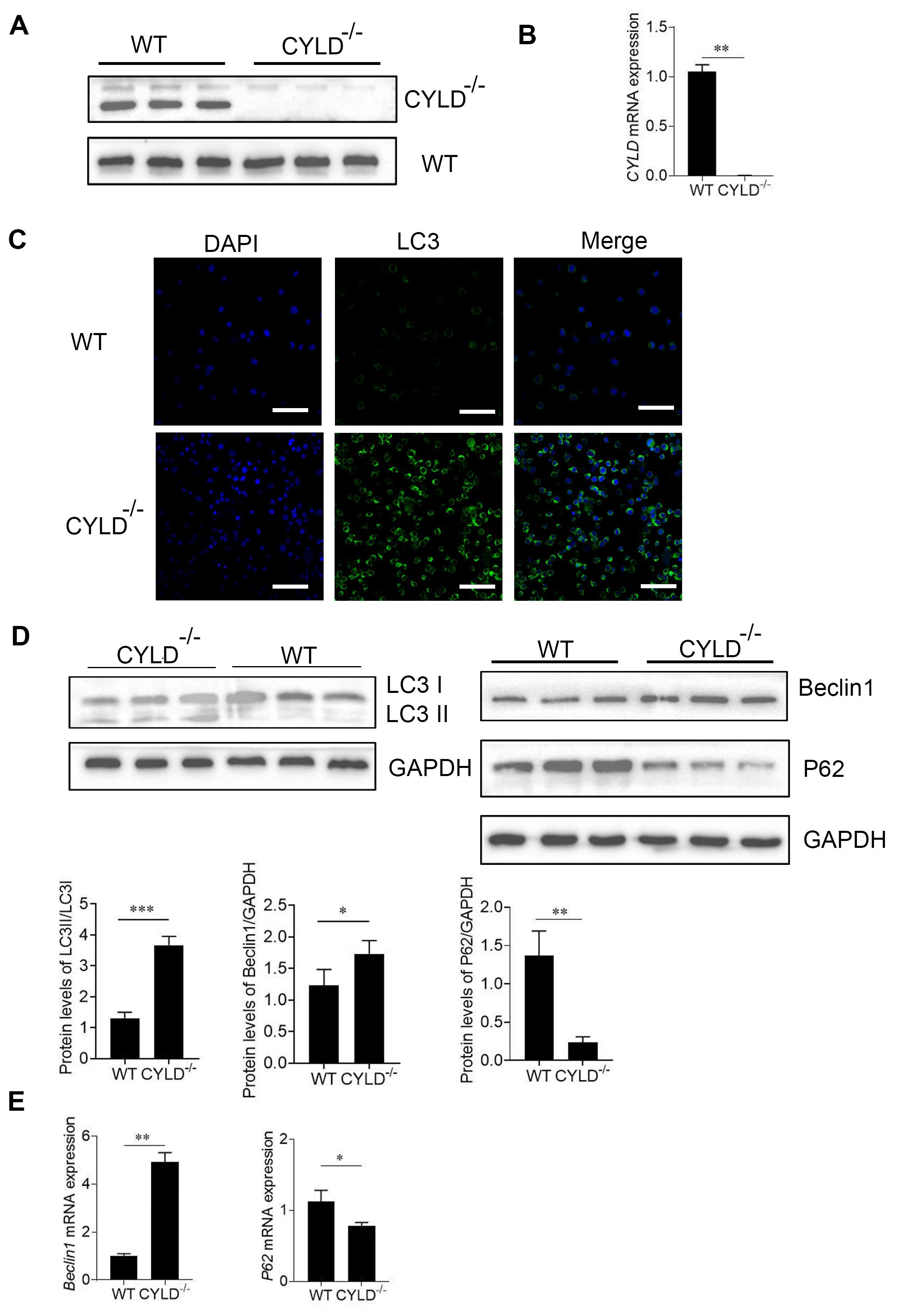
To investigate the association between CYLD and autophagy, the CYLD gene was knocked out in Jurkat cells, and these cells were subsequently analyzed using Western blotting and RT-PCR.The results showed that CYLD knockout was successful (A,B). At the same time, we briefly verified that after CYLD knockout, there were no significant differences in growth rate and cell size compared to the control group cells (Figure S1A,B). During autophagy, LC3‑I is converted to LC3‑II by lipidation through a ubiquitin‑like system which involves Atg3 and Atg7; therefore, LC3 is associated with autophagic vesicles, and the presence of LC3 in autophagosomes is the indicator of autophagy [
23]. Firstly, there were no significant differences in LC3 expression levels by western blot between the CYLD gene knockout group and the control group in the basal state (Figure S1C). Immunofluorescence shows the number of cells with positive GFP LC3 puncta was significantly increased when CYLD was knocked out (C). At the time, we tested some indicators related to autophagy indicators including LC3, Beclin1 and P62. Western Blot and RT-PCR results indicate the expression of Beclin1 was significantly increased at the protein and mRNA levels in CYLD knockout cells. And the expression of P62 was significantly decreased in the protein and mRNA levels in CYLD knockout cells. The LC3 II conversion was higher as compared with the control (D,E). These data suggest CYLD deletion may induce autophagy in Jurkat cells.
. CYLD deletion induces autophagy in Jurkat cells. (<b>A</b>) The protein level of CYLD gene was evaluated by immunoblotting analysis. (<b>B</b>) RT-PCR analysis for mRNA expression of CYLD in control and CYLD knockout Jurkat cells and expressed as a ratio to <i>GAPDH</i>. (<b>C</b>) The expression of GFP-LC3 positive autophagosomes was examined by immunofluorescence analysis. GFP-LC3; green and DAPI; blue. (<b>D</b>) The protein level of LC3, Beclin1 and P62 were evaluated by immunoblotting analysis. Under lanes is a quantization of the Western blot results. (<b>E</b>) RT-PCR analysis for mRNA expression of <i>Beclin1</i> and <i>P62</i> in the two groups. The data shown represent three independent experiments with similar results. <sup>∗</sup>, <i>p</i> < 0.05; <sup>∗∗</sup>, <i>p</i> < 0.01; <sup>∗∗∗</sup>, <i>p</i> < 0.001.
Previous research has shown AMP-dependent protein kinase (AMPK) is an important protein kinase that regulates changes in cellular sensory energy [
24]. It can regulate autophagy by mammalian targets of rapamycin (mTOR) and degrade the intracellular substance to produce energy [
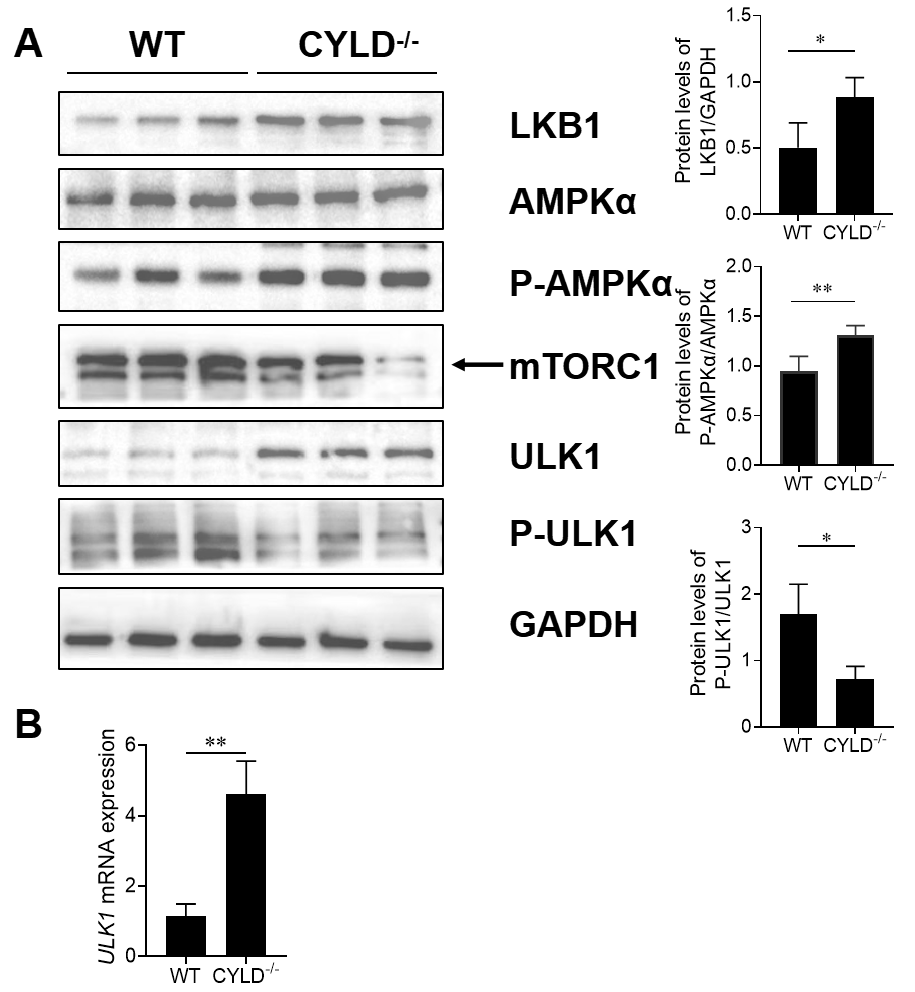
25]. To determine whether CYLD deletion regulates autophagy through AMPKα/mTOR/ULK1 pathway, we tested AMPKα, mTOR and ULK1 activity in host cells. Compared with the control, increased phosphorylation levels of AMPKα and its downstream target ULK1 and concurrent reduction in levels of mTOR were typically observed in the CYLD knockout cells undergoing autophagy. The phosphorylation levels of ULK1 were down-regulated. While total AMPKα do not change (A). RT-PCR demonstrates that
ULK1 was up-regulated in CYLD knockout cells (B). LKB1 is the upstream kinase necessary for activation of AMPK under low-energy conditions, so AMPKα activation requires LKB1. Western blot showed LKB1 was up-regulated in CYLD knockout cells. AMPKα/mTOR/ULK1 signaling pathway was activated. These findings show that CYLD deletion induces autophagy through the AMPKα/mTOR/ULK1 signaling pathway.
. Effects of CYLD gene on LKB1/AMPKα/ULK1 signaling pathway. (<b>A</b>) The protein level of LKB1, total AMPKα, AMPK phosphorylation (P-AMPK), mTORC1, total ULK1 and ULK1 phosphorylation (P-ULK1) were evaluated by immunoblotting analysis. (<b>B</b>) RT-PCR analysis for mRNA expression of <i>ULK1</i> in control and CYLD knockout Jurkat cells. The data shown represent three independent experiments with similar results. <sup>∗</sup>, <i>p</i> < 0.05; <sup>∗∗</sup>, <i>p</i> < 0.01.
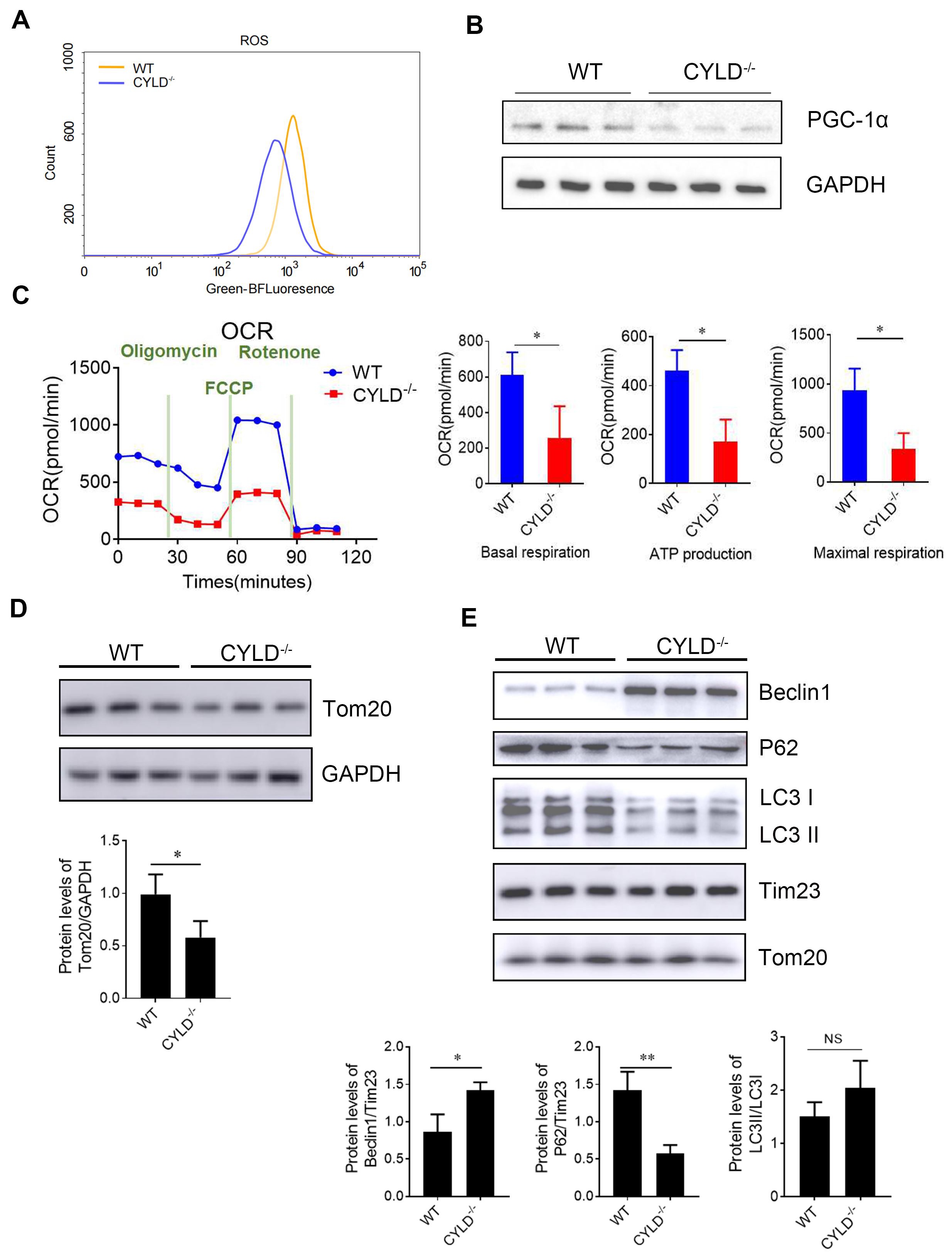
It has been noted that activation of AMPKα can reduce reactive oxygen species (ROS) production in mitochondria [
26]. While mitochondria have been considered a common venue of ROS production [
27,
28]. Previous results indicate that the deletion of CYLD activates the AMPKα pathway. So, we tested ROS production by flow cytometry. Results showed that CYLD deletion reduced ROS production (A). This suggests that CYLD deletion may affect mitochondrial function. Mitochondrial dysfunction may impair cellular energy supply and increase oxidative stress. Peroxisome proliferator-activated receptor γ coactivator-α1 (PGC-1α) plays a key role in the biosynthesis function of mitochondria [
29]. Western blot showed PGC1-α was down-regulated when CYLD was knocked out. At the same time, results showed that mitochondrial respiratory function decreased after CYLD deletion by seahorse bioscience (XFe24) (C). The result showed that CYLD deletion reduced the energy metabolism of mitochondria. The above results indicate that CYLD deletion affects mitochondrial function.
Mitochondrial autophagy can occur under stressful conditions such as ROS production, poor nutrition or cellular aging. Then, we tested the outer membrane proteins Tom20. Western blot results showed that the protein levels of Tom20 and were decreased (D). Autophagy machinery is also utilized to remove damaged mitochondria, and thus, this process is termed mitophagy. We extracted mitochondrial protein to detect autophagy-related indicators. Western blot results showed that the protein level of Beclin1 was up-regulated compared with the control. The LC3 II conversion was higher (E). These findings show that mitophagy is increased. Autophagy induced by CYLD deletion is consistent with mitochondrial autophagy. These results suggest that CYLD deletion affects mitochondrial function and promotes mitophagy to improve or eliminate damaged mitochondria.
. CYLD deletion affects mitochondrial function and promotes mitochondrial turnover (mitophagy). (<b>A</b>) Measurement of intracellular reactive oxygen species (ROS) level by flow cytometry. (<b>B</b>) The protein level of PGC1-α was evaluated by immunoblotting analysis. (<b>C</b>) Measurement of Mitochondrial respiration by Seahorse XF Analyzer. (<b>D</b>) The protein level of Tom20 in the host cells by immunoblotting analysis. (<b>E</b>) The mitochondrial protein level of indicators related to autophagy indicators, including Beclin1, P62 and LC3, by immunoblotting analysis. The data shown represent three independent experiments with similar results. <sup>∗</sup>, <i>p</i> < 0.05; <sup>∗∗</sup>, <i>p</i> < 0.01.
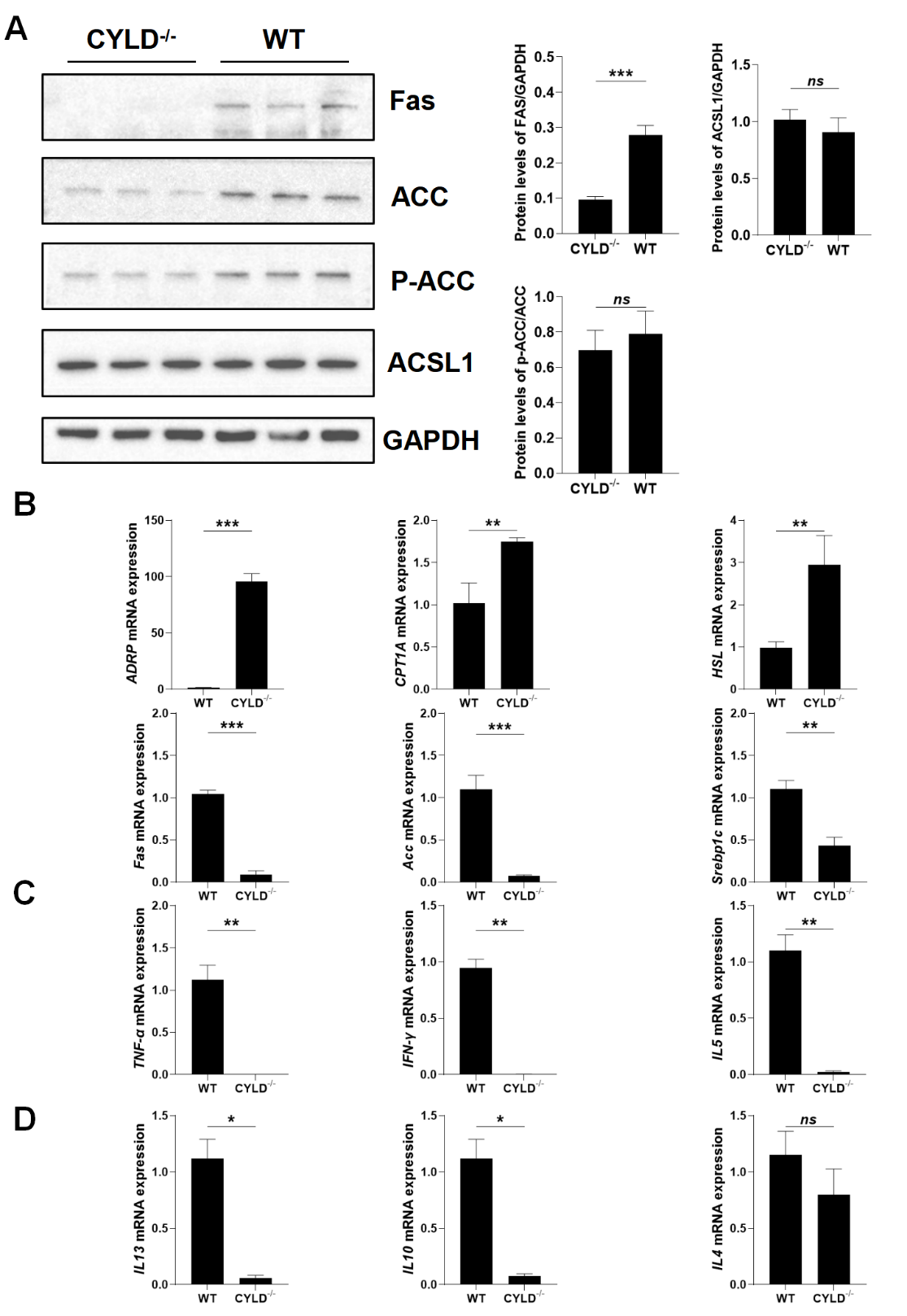
Studies have shown that the LKB1/AMPKα signaling pathway plays a very important role in lipid metabolism. During energy stress, AMPK directly phosphorylates key factors involved in multiple pathways to restore energy balance [
30]. AMPKα activation inactivates the rate-limiting enzymes associated with lipolysis [
31]. It has been reported that autophagy can occur in other metabolic effects, such as fat synthesis [
32]. So, we tested the key fatty acid synthesis enzymes downstream of AMPKα, such as ACC, FAS, and SREBP1c. Western blot and RT-PCR results showed that the expression of ACC, FAS, P-ACC and SREBP1c were down-regulated in the protein and mRNA levels when CYLD was knocked out. At the same time, the expression of some lipolytic rate-limiting enzymes, such as hormone-sensitive triglyceride lipase (HSL) and ADRP were up-regulated (A,B). Also the protein and mRNA expression of CPT1A was raised in CYLD deletion cells (Figure S2A and B). This suggests that CYLD deletion not only affects mitochondrial function but also affects cell metabolism in addition to regulating autophagy.
. CYLD affects the overall cell metabolism and T cell function. (<b>A</b>) The protein level of SREBP1c, ACC phosphorylation (p-ACC), total ACC, FAS, and ACSL1 by immunoblotting analysis. (<b>B</b>) RT-PCR analysis for mRNA expression of synthesis and lipolytic rate-limiting enzyme FAS, ACC, SREBP1c, ADRP, HSL and CPT1A. (<b>C</b>) RT-PCR analysis for mRNA expression of proinflammatory factor <i>TNF-α</i>, <i>IFN-γ</i> and <i>IL-5</i>. (<b>D</b>) RT-PCR analysis for mRNA expression of anti-flammatory factor <i>IL-4</i>, <i>IL-10</i> and <i>IL-13</i>. Data are represented as mean ± SD (<i>n</i> = 3). <sup>∗</sup>, <i>p</i> < 0.05; <sup>∗∗</sup>, <i>p</i> < 0.01; <sup>∗∗∗</sup>, <i>p</i> < 0.001.
While it is reported that CYLD is transcriptionally upregulated by IFNγ-induced transcription factor IRF1 as a deubiquitinating enzyme [
33]. And in patients with ulcerative colitis, the concentration of active IL-18 was inversely correlated with CYLD expression. CYLD deficiency in mice resulted in elevated levels of active IL-18 and severe colonic inflammation following Citrobacter rodentium infection [
34]. We also tested the expression of related inflammatory factors by real-time PCR and ELISA. The results showed that the expression of related inflammatory factors was decreased when CYLD was knocked out (C,D and Figure S3A). We speculated that CYLD deletion affected the ability of T cells to secrete inflammatory factors.
4. Discussion
Ubiquitylation and deubiquitylation are established posttranslational mechanisms for regulating immune responses, as well as the development and activation of immune cells [
35]. CYLD plays a fundamental role in regulating T cell development and activation. Cyld-deficient mice show delayed thymocyte development due to a constitutively K48-ubiquitylated and degraded LCK protein [
9]. A large number of reports indicate that the development and activation of T lymphocytes are related to autophagy and cellular metabolism [
36,
37,
38,
39]. Previous studies have reported that overexpression of CYLD gene can inhibit autophagy in cancer cells, but CYLD has no specific study on the specific mechanism of autophagy regulation and its effect on cell metabolism in T cells. This study demonstrates that CYLD deletion promotes autophagy through the AMPKα/mTOR/ULK1 signaling pathway and that CYLD deletion activates the AMPKα pathway to improve mitochondrial function and cellular lipid metabolism. This shows a additional mechanism for the interaction of CYLD on autophagy and lipid metabolism, pointing out the new role of CYLD in autophagy and lipid metabolism.
In our study, we found that the deletion of CYLD promoted autophagy in Jurkat cells. Previous studies have shown that CYLD is involved in the process of autophagy [
19], so we knocked out CYLD in the Jurkat cells to explore the effect of CYLD on autophagy, and the results showed that autophagy was promoted when CYLD was absent.
Numerous studies have shown that the AMPKα/mTOR/ULK1 signaling pathway plays a major role in the regulation of autophagy [
19]. As an autophagy-initiating kinase, the mechanism of ULK1 regulation is central to understanding autophagy regulation. AMPK senses cellular energy status and activates ULK1 kinase by a coordinated cascade. Studies have demonstrated that under glucose starvation, the activated AMPK inhibits mTORC1 to reduce the phosphorylation of ULK1, leading to ULK1/AMPK interaction [
40]. Phosphorylation of AMPKα activates ULK1 kinase, which ultimately leads to autophagy induction [
21]. In our study, we examined the upstream and downstream key molecules of the AMPKα signaling pathway. The results showed that in CYLD knockout cells, LKB1, AMPK phosphorylation, ULK1 expression was increased, and mTOR expression was decreased. These results indicate that CYLD may regulate the occurrence of autophagy through AMPKα/mTOR/ULK1 signaling pathway.
While the phosphorylation of ULK1 Ser555 by AMPKα is critical to autophagy, cell survival and mitochondrial balance [
40]. Activation of AMPKα leads to reduced production of ROS, while mitochondria have been considered as a common venue of oxygen-free radical production. When autophagy occurs to remove damaged mitochondria, it is called mitophagy. Our previous data showed that CYLD activated AMPKα pathway, so we detected mitochondrial function. We found CYLD deletion affected biosynthesis function, respiratory function, and the ability to produce ROS of mitochondrial. At the same time, we tested Tom20 and Tim23 by western blot. We found CYLD deletion-induced membrane protein expression was decreased. This suggests that CYLD deletion may promote mitochondrial autophagy, so we extract mitochondrial protein to examine indicators related to autophagy. We found that mitochondria autophagy was increased. These explained CYLD deletion affected the function of mitochondria. Mitophagy is activated to improve mitochondrial function.
It is well known that AMPK is an important cellular energy regulator [
41] and plays an important role in fat production, fatty acid decomposition and oxidation. Acetyl-CoA carboxylase (ACC) is a key regulator of fatty acid synthesis and an essential substrate for AMPK. Studies have shown that fatty acid synthesis in rat hepatocytes is inhibited during mild heat stress (42°C) (AMPK is partially activated). While our data showed that the expression of ACC and the phosphorylation levels were both decreased. That means the activity of ACC was decreased. The mechanism of AMPKα promoting β-oxidation of fatty acid is that the production of propionyl-CoA is decreased after ACC is inhibited. Since propionyl-CoA is an inhibitor of carnitine fatty acyltransferase (CPT-1A), our data showed that CPT-1A expression was increased at the mRNA level, which is the acyl-CoA from the cytosol into the mitochondria, thus starting for β-oxidation. At the same time, this is consistent with the increase in ATP in mitochondria when CYLD is absent (complement). Our data also showed that fatty acid synthase FAS and SREBP1c were decreased significantly, and some lipolysis rate-limiting enzymes such as HSL and ADRP were increased. These results indicate that CYLD deletion may attenuate lipid synthesis via the AMPK pathway and improve cellular metabolism. However, our experiment also has limitations. There are multiple signaling pathways that affect cell metabolism. We did not use AMPK inhibitors to confirm that the pathway through which CYLD affects cell lipid metabolism is indeed through the AMPK signaling pathway. This is worth further experimentation.
Our study used CYLD knockout model in Jurkat cells. We found that CYLD deletion promoted autophagy, affected mitochondrial function, promoted mitochondrial autophagy, and improved cellular metabolism through the AMPK pathway. Our findings offer new insights into the interaction between autophagy and, lipid synthesis and mitochondria in T cells. But there are still some complicated relationships between them. It requires further exploration.
Supplementary Materials
The following supporting information can be found at: https://www.sciepublish.com/article/pii/399. Figure S1. Two groups of cells in basic conditions. (A). CCK-8 assay to determine the proliferation of Jurkat cells and CYLD knock Jurkat T cells. (B) The image of Jurkat cells and CYLD knock Jurkat T cells (20x). (C) The protein level of LC3 were evaluated by immunoblotting analysis in basal conditions. (D) The mRNA expression level of GAPDH by RT-PCR. The data shown represent three independent experiments with similar results; Figure S2. The protein level of CPT1A; Figure S3. CYLD deficiency decreased inflammatory factors. (A,B). The level of inflammatory factors in the supernatant was measured by ELISA analysis. The data shown represent three independent experiments with similar results.
Author Contributions
H.W. and L.Y. wrote the manuscript; H.W., L.Y., and J.Y. contributed conception and design of the study. L.Y., G.G., J.G. and Y.Z. contributed to the experiments. Y.L. generated and supplied CYLD−/− Jurkat cells. Y.N. provided expertise and advice. H.W. oversaw the project. All authors contributed to the manuscript revision, and have read and approved the submitted version.
Ethics Statement
Not applicable.
Informed Consent Statement.
Not applicable.
Data Availability Statement
Data will be made available on request.
Funding
This work was funded by the National Natural Science Foundation of China (Grant No. U1804190) and the 111 Project (No. D20036).
Declaration of Competing Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1.
Haas R, Cucchi D, Smith J, Pucino V, Macdougall CE, Mauro C. Intermediates of Metabolism: From Bystanders to Signalling Molecules.
Trends Biochem. Sci. 2016,
41, 460–471.
[Google Scholar]
2.
Liu H, Zeng L, Pan M, Huang L, Li H, Liu M, et al. Bcl-3 regulates T cell function through energy metabolism.
BMC Immunol. 2023,
24, 35.
[Google Scholar]
3.
Zhang X, Liu J, Cao X. Metabolic control of T-cell immunity via epigenetic mechanisms.
Cell Mol. Immunol. 2018,
15, 203–205.
[Google Scholar]
4.
Wang R, Green DR. Metabolic checkpoints in activated T cells.
Nat. Immunol. 2012,
13, 907–915.
[Google Scholar]
5.
Mehta MM, Weinberg SE, Chandel NS. Mitochondrial control of immunity: beyond ATP.
Nat. Rev. Immunol. 2017,
17, 608–620.
[Google Scholar]
6.
Buck MD, O’Sullivan D, Geltink RIK, Curtis JD, Chang CH, Sanin DE, et al. Mitochondrial Dynamics Controls T Cell Fate through Metabolic Programming.
Cell 2016,
166, 63–76.
[Google Scholar]
7.
Chiticariu E, Regamey A, Huber M, Hohl D. CENPV Is a CYLD-Interacting Molecule Regulating Ciliary Acetylated alpha-Tubulin.
J. Investig. Dermatol. 2020,
140, 66–74 e4.
[Google Scholar]
8.
Mathis JB, Lai Y, Qu C, Janicki SJ, Cui T. CYLD-mediated signaling and diseases.
Curr. Drug Targets 2015,
16, 284–294.
[Google Scholar]
9.
Reiley WW, Zhang M, Jin W, Losiewicz M, Donohue KB, Norbury CC, et al. Regulation of T cell development by the deubiquitinating enzyme CYLD.
Nat. Immunol. 2006,
7, 411–417.
[Google Scholar]
10.
Lee BC, Miyata M, Lim JH, Li JD. Deubiquitinase CYLD acts as a negative regulator for bacterium NTHi-induced inflammation by suppressing K63-linked ubiquitination of MyD88.
Proc. Natl. Acad. Sci. USA 2016,
113, E165–E171.
[Google Scholar]
11.
Zhang LM, Zhou JJ, Luo CL. CYLD suppression enhances the pro-inflammatory effects and hyperproliferation of rheumatoid arthritis fibroblast-like synoviocytes by enhancing NF-kappaB activation.
Arthritis Res. Ther. 2018,
20, 219.
[Google Scholar]
12.
Lork M, Verhelst K, Beyaert R. CYLD, A20 and OTULIN deubiquitinases in NF-kappaB signaling and cell death: so similar, yet so different.
Cell Death Differ. 2017,
24, 1172–1183.
[Google Scholar]
13.
Massoumi R, Chmielarska K, Hennecke K, Pfeifer A, Fässler R. Cyld inhibits tumor cell proliferation by blocking Bcl-3-dependent NF-kappaB signaling.
Cell 2006,
125, 665–677.
[Google Scholar]
14.
Fernández-Majada V, Welz PS, Ermolaeva MA, Schell M, Adam A, Dietlein F, et al. The tumour suppressor CYLD regulates the p53 DNA damage response.
Nat. Commun. 2016,
7, 12508.
[Google Scholar]
15.
Hellerbrand C, Bumes E, Bataille F, Dietmaier W, Massoumi R, Bosserhoff AK. Reduced expression of CYLD in human colon and hepatocellular carcinomas.
Carcinogenesis 2007,
28, 21–27.
[Google Scholar]
16.
Pseftogas A, Xanthopoulos K, Siasiaridis A, Poutahidis T, Gonidas C, Tsingotjidou A, et al. Inactivation of the Tumor Suppressor CYLD Sensitizes Mice to Breast Cancer Development.
Anticancer Res. 2024,
44, 1885–1894.
[Google Scholar]
17.
Wang J, Niu Z, Shi Y, Gao C, Wang X, Han J, et al. Bcl-3, induced by Tax and HTLV-1, inhibits NF-kappaB activation and promotes autophagy.
Cell Signal 2013,
25, 2797–2804.
[Google Scholar]
18.
Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research.
Cell 2010,
140, 313–326.
[Google Scholar]
19.
Yin L, Liu S, Li C, Ding S, Bi D, Niu Z, et al. CYLD downregulates Living and synergistically improves gemcitabine chemosensitivity and decreases migratory/invasive potential in bladder cancer: the effect is autophagy-associated.
Tumour Biol. 2016,
37, 12731–12742.
[Google Scholar]
20.
Bonapace L, Bornhauser BC, Schmitz M, Cario G, Ziegler U, Niggli FK, et al. Induction of autophagy-dependent necroptosis is required for childhood acute lymphoblastic leukemia cells to overcome glucocorticoid resistance.
J. Clin. Investig. 2010,
120, 1310–1323.
[Google Scholar]
21.
Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1.
Nat. Cell Biol. 2011,
13, 132–141.
[Google Scholar]
22.
Li X, Liu J, Lu Q, Ren D, Sun X, Rousselle T, et al. AMPK: A therapeutic target of heart failure-not only metabolism regulation.
Biosci. Rep. 2019,
39, BSR20181767.
[Google Scholar]
23.
Hou Y, Fu J, Sun S, Jin Y, Wang X, Zhang L. BDE-209 induces autophagy and apoptosis via IRE1alpha/Akt/mTOR signaling pathway in human umbilical vein endothelial cells.
Environ. Pollut. 2019,
253, 429–438.
[Google Scholar]
24.
Lee SB, Kim JJ, Han SA, Fan Y, Guo LS, Aziz K, Nowsheen S, et al. The AMPK-Parkin axis negatively regulates necroptosis and tumorigenesis by inhibiting the necrosome.
Nat. Cell. Biol. 2019,
21, 940–951.
[Google Scholar]
25.
Chen X, Luo Y, Wang M, Sun L, Huang K, Li Y, et al. Wuhu Decoction regulates dendritic cell autophagy in the treatment of respiratory syncytial virus (RSV)-induced mouse asthma by AMPK/ULK1 signaling pathway.
Med. Sci. Monit. 2019,
25, 5389–5400.
[Google Scholar]
26.
Guan J, Lin H, Xie M, Huang M, Zhang D, Ma S, et al. Higenamine exerts an antispasmodic effect on cold-induced vasoconstriction by regulating the PI3K/Akt, ROS/alpha2C-AR and PTK9 pathways independently of the AMPK/eNOS/NO axis.
Exp. Ther. Med. 2019,
18, 1299–1308.
[Google Scholar]
27.
Mohsin AA, Chen Q, Quan N, Rousselle T, Maceyka MW, Samidurai A, et al. Mitochondrial complex I inhibition by metformin limits reperfusion injury.
J. Pharmacol. Exp. Ther. 2019,
369, 282–290.
[Google Scholar]
28.
Ross T, Szczepanek K, Bowler E, Hu Y, Larner A, Lesnefsky EJ, et al. Reverse electron flow-mediated ROS generation in ischemia-damaged mitochondria: role of complex I inhibition vs. depolarization of inner mitochondrial membrane.
Biochim. Biophys. Acta 2013,
1830, 4537–4542.
[Google Scholar]
29.
Li L, Pan R, Li R, Niemann B, Aurich AC, Chen Y, et al. Mitochondrial biogenesis and peroxisome proliferator-activated receptor-gamma coactivator-1alpha (PGC-1alpha) deacetylation by physical activity: intact adipocytokine signaling is required.
Diabetes 2011,
60, 157–167.
[Google Scholar]
30.
Herzig S, Shaw RJ. AMPK: guardian of metabolism and mitochondrial homeostasis.
Nat. Rev. Mol. Cell Biol. 2018,
19, 121–135.
[Google Scholar]
31.
Seo MS, Kim JH, Kim HJ, Chang KC, Park SW. Honokiol activates the LKB1-AMPK signalling pathway and attenuates the lipid accumulation in hepatocytes.
Toxicol. Appl. Pharmacol. 2015,
284, 113–124.
[Google Scholar]
32.
Deng J, Guo Y, Yuan F, Chen S, Yin H, Jiang X, et al. Autophagy inhibition prevents glucocorticoid-increased adiposity via suppressing BAT whitening.
Autophagy 2020,
16, 451–465.
[Google Scholar]
33.
Deng B, Wang J, Yang T, Deng Z, Yuan J, Zhang B, et al. TNF and IFNgamma-induced cell death requires IRF1 and ELAVL1 to promote CASP8 expression.
J. Cell Biol. 2024,
223, e202305026.
[Google Scholar]
34.
Mukherjee S, Kumar R, Tsakem Lenou E, Basrur V, Kontoyiannis DL, Ioakeimidis F, et al. Deubiquitination of NLRP6 inflammasome by Cyld critically regulates intestinal inflammation.
Nat. Immunol. 2020,
21, 626–635.
[Google Scholar]
35.
Thuille N, Wachowicz K, Hermann-Kleiter N, Kaminski S, Fresser F, Lutz-Nicoladoni C, et al. PKCtheta/beta and CYLD are antagonistic partners in the NF-kappaB and NFAT transactivation pathways in primary mouse CD3+ T lymphocytes.
PLoS ONE 2013,
8, e53709.
[Google Scholar]
36.
DeVorkin L, Pavey N, Carleton G, Comber A, Ho C, Lim J, et al. Autophagy regulation of metabolism is required for CD8
+ T cell anti-tumor immunity.
Cell Rep. 2019,
27, 502–513 e5.
[Google Scholar]
37.
Dowling SD, Macian F. Autophagy and T cell metabolism.
Cancer Lett. 2018,
419, 20–26.
[Google Scholar]
38.
Mocholi E, Dowling SD, Botbol Y, Gruber RC, Ray AK, Vastert S, et al. Autophagy is a tolerance-avoidance mechanism that modulates TCR-mediated signalling and cell metabolism to prevent induction of T cell anergy.
Cell Rep. 2018,
24, 1136–1150.
[Google Scholar]
39.
Yang G, Song W, Postoak JL, Chen J, Martinez J, Zhang J, et al. Autophagy-related protein PIK3C3/VPS34 controls T cell metabolism and function.
Autophagy 2020,
1, 1–12.
[Google Scholar]
40.
Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy.
Science 2011,
331, 456–561.
[Google Scholar]
41.
Foretz M, Guigas B, Viollet B. Understanding the glucoregulatory mechanisms of metformin in type 2 diabetes mellitus.
Nat. Rev. Endocrinol. 2019,
15, 569–589.
[Google Scholar]