Background: The strains of COVID-19 are constantly mutating, and the effectiveness of Chinese inactivated vaccines against the COVID-19 Delta variant has not been described clearly. Methods: The clinical data of patients with the COVID-19 Delta variant in the 2021 Nanjing outbreak were retrospectively reviewed. Results: There were 212 patients with the COVID-19 Delta variant (unvaccinated, n = 56, 26.42%; vaccinated, n = 156, 73.58%) included in our cohort study. The median age was 45.5 (38, 53) years old. Eighty-seven subjects (41.04%) were airport staff, and 94 patients (44.34%) in 32 families were infected. There were 53 (25.00%) and 103 (48.58%) cases with one-dose and two-dose vaccination, respectively, and 55 (25.94%), 147 (69.34%) and 10 (4.72%) had mild, moderate and severe symptoms, respectively. The duration of viral shedding, or viral shedding time (VST), was significantly longer in unvaccinated individuals compared to vaccinated individuals (p = 0.0008). Moreover, the duration was significantly longer in patients who received one vaccine dose than those who received two doses (p < 0.0001). The mild patients had significantly shorter VSTs than the moderate subjects (p < 0.0001). Disease severity and vaccination dose were independent predictors for VST by Cox regression models. Conclusions: These results suggest that two-dose vaccination could reduce VST in patients with the COVID-19 Delta variant. Chinese inactivated vaccines may decrease the disease severity of cases with the COVID-19 Delta variant.

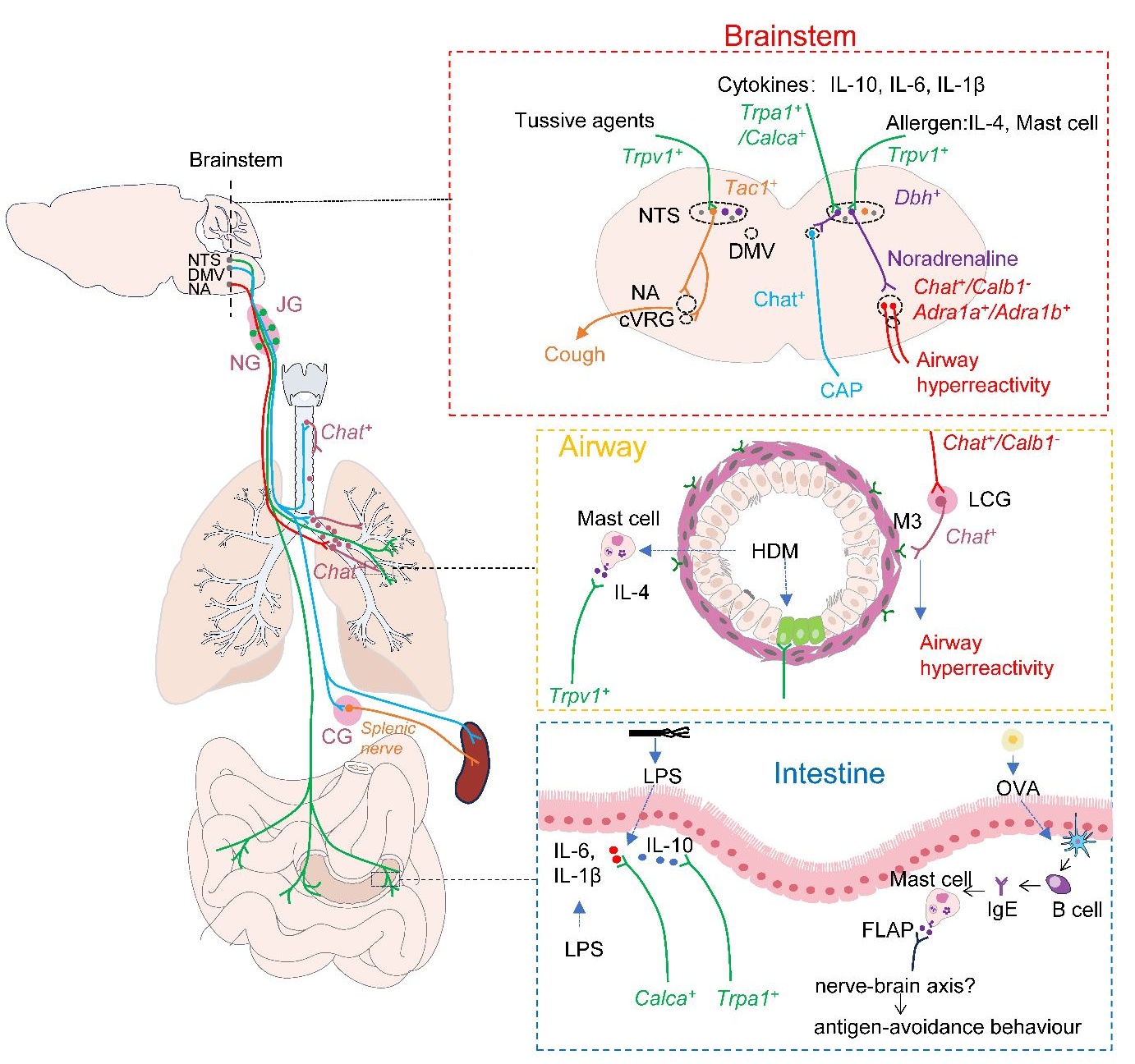
The nucleus of the solitary tract (NTS) is the primary hub for sensing and integrating respiratory information. It integrates input from the vagus and glossopharyngeal nerve. It interacts with other brainstem nuclei, such as the nucleus ambiguus (NA) and the dorsal motor nucleus of the vagus (DMV), to transmit information and initiate a neuroreflex response to respiratory stimuli. In a recent issue of the journal Nature, Su et al. demonstrated that Dbh+ neurons in the NTS can receive signals from vagal Trpv1+ sensory neurons that sense allergen−induced IL−4 production in mast cells and pass the signal to Chat+ neurons in the NA by releasing norepinephrine. Subsequently, NA Chat+ neurons drive allergen−induced airway hyperresponsiveness by projecting onto cholinergic pulmonary ganglia in the lungs. This study not only provides new insights into the regulation of allergen−induced airway hyperresponsiveness by lung−vagus–brainstem interoceptive circuit but also provides us with new strategies to combat asthma.
The aim of the study was to compare Hypoxic Ventilatory Response (HVR) of sleep apnea in Uygur patients stemming from higher altitude and Chinese Han patients from sea level. 276 subjects with or without snoring from the Karamay community were recruited. 226 subjects (n = 71 Han OSA patients, n = 75 Uygur OSA patients, n = 52 for Uygur control subjects without OSA, n = 28 Han control subjects without OSA) were matched for age and gender. All patients were assessed via polysomnography (PSG). Lung function was assessed. Apnea-hypopnea index (AHI), mean SaO2 (MSaO2%), lowest SaO2 (LSaO2%), the number of desaturations ≥4% per hour (ODI4), FEV1/FVC ratio, HVR, △VE/△SaO2 and the pulse responses to hypoxia changes (ΔPulse/ΔSaO2) were calculated. A multiple logistic regression using a binary outcome for HVR was applied. (1) In control subjects without OSA, those living at high altitude (Uygur) had a lower HVR than control subjects living at sea level (Han) [−0.35L·min−1 per %SpO2(−0.49 to−0.20 L·min−1 per %SpO2) vs.−0.44 L·min−1 per %SpO2(−0.55 to −0.21 L·min−1 per %SpO2)]. (2) Compared to patients with OSA living at sea level (Han), those OSA patients living at high altitude (Uygur) had a higher neck circumference [43 cm (range 39–45 cm) vs. 42 cm (41–46) cm], higher abdominal circumference [110 cm (102–120 cm) vs. 101 cm (98–111 cm], higher LSaO2 [81% (72–85%) vs. 76% (68–81%)], lower AHI [26 events/h (16–43 events/h) vs. 36 events/h (24–62 events/h)] and lower ODI4 [15/h (7–29/h) vs. 37/h (20–54/h)]. (3) Considering patients with mild OSA, those who lived at high altitude (Uygur) had a weaker HVR compared to Han patients [−0.31 L·min−1 per %SpO2(−0.42 to −0.20 L·min−1 per %SpO2) vs.−0.47 L·min−1 per %SpO2(−0.59 to −0.21 L·min−1 per %SpO2)]. However, in moderate and severe OSA the difference in HVR between people living at high and low altitudes was not significant. In people living at high altitude (Uygur) compared to sea level (Han), HVR is weaker both in control subjects and those with mild OSA, but this difference between populations living at different altitudes in those with moderate and severe OSA is not obvious.

The conducting airways of the respiratory system play a crucial role in filtering, humidifying, and directing air into the lungs. Among the specialized cell types within these airways, airway serous cells are notable for their secretion of watery, protein-rich fluids and enzymes, which contribute to maintaining airway surface liquid homeostasis and defending against pathogens. However, the distribution and abundance of serous cells across different species in the conducting airways remain poorly understood. In this study, we addressed this gap by investigating the spatial distribution of the airway serous cell-specific marker BPI fold containing family A member 1 (BPIFA1) in humans, pigs, and mice. Our findings demonstrate significant variations in the distribution and abundance of serous cells among these species, potentially reflecting their different respiratory anatomy and evolutionary adaptations to diverse environmental challenges and respiratory demands. In humans and pigs, airway serous cells are predominantly found in the submucosal glands of the trachea and segmental bronchi, frequently overlapping with lysozyme-positive secretory cells. In contrast, rodents like mice exhibit a distinct pattern where serous cells are scarce in submucosal glands. Instead, rodent serous cells are primarily located at the epithelial surface from the trachea to the main bronchi, where many co-express the Club cell-specific protein SCGB1A1. The abundance of serous cells diminishes progressively in the intrapulmonary airways. Given that rodent models are widely utilized in respiratory research, understanding anatomical and cellular differences in airway serous cells is critical for interpreting experimental outcomes and translating findings to human respiratory diseases and therapeutic strategies. This comparative analysis enhances our understanding of airway biology across species and informs the selection and interpretation of animal models in respiratory studies.

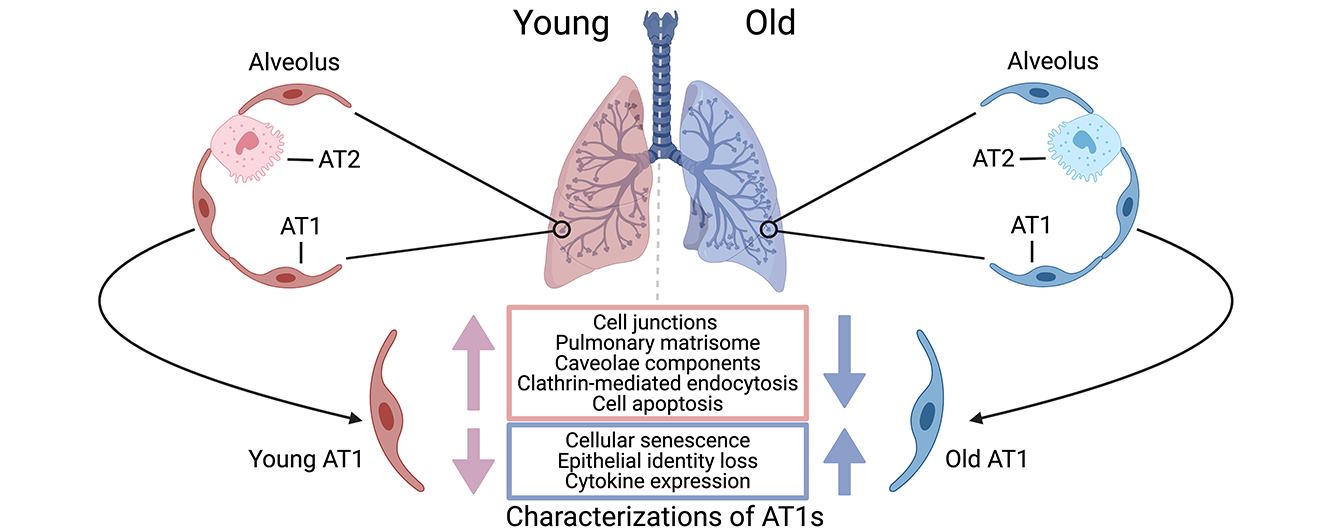
Human alveolar type I (AT1) cells are specialized epithelial cells that line the alveoli in the lungs where gas exchange occurs. The primary function of AT1 cells is not only to facilitate efficient gas exchange between the air and the blood in the lungs, but also to contribute to the structural integrity of the alveoli to maintain lung function and homeostasis. Aging has notable effects on the structure, function, and regenerative capacity of human AT1 cells. However, our understanding of the molecular mechanisms driving these age-related changes in AT1 cells remains limited. Leveraging a recent single-cell transcriptomics dataset we generated on healthy human lungs, we identified a series of significant molecular alterations in AT1 cells from aged lungs. Notably, the aged AT1 cells exhibited increased cellular senescence and chemokine gene expression, alongside diminished epithelial features such as decreases in cell junctions, endocytosis, and pulmonary matrisome gene expression. Gene set analyses also indicated that aged AT1 cells were resistant to apoptosis, a crucial mechanism for turnover and renewal of AT1 cells, thereby ensuring alveolar integrity and function. Further research on these alterations is imperative to fully elucidate the impact on AT1 cells and is indispensable for developing effective therapies to preserve lung function and promote healthy aging.
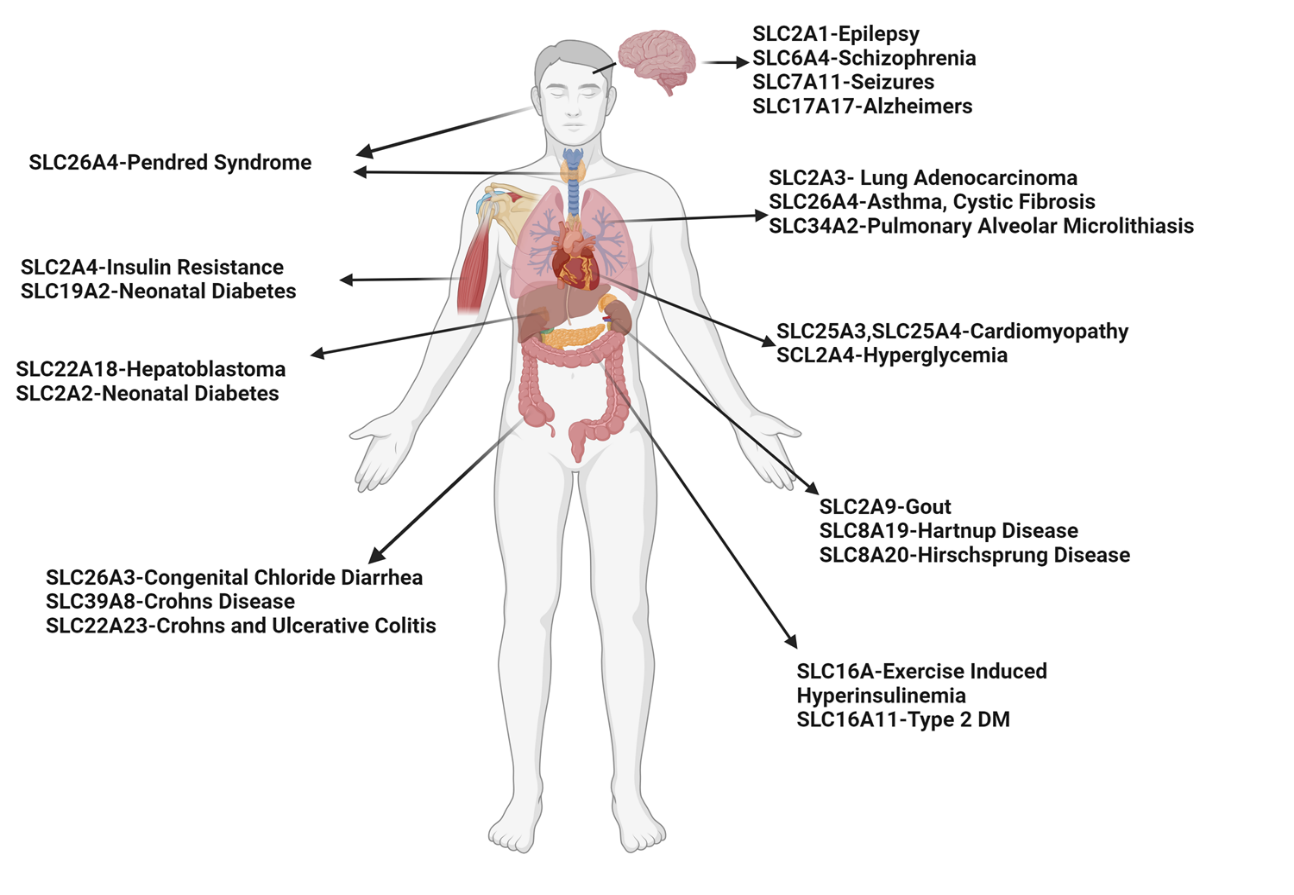
Asthma is a prevalent respiratory condition with multifaceted pathomechanisms, presenting challenges for therapeutic development. The SLC (Solute Carrier) gene family, encompassing diverse membrane transport proteins, plays pivotal roles in various human diseases by facilitating solute movement across biological membranes. These solutes include ions, sugars, amino acids, neurotransmitters, and drugs. Mutations in these ion channels have been associated with numerous disorders, underscoring the significance of SLC gene families in physiological processes. Among these, the SLC26A4 gene encodes pendrin, an anion exchange protein involved in transmembrane transport of chloride, iodide, and bicarbonate. Mutations in SLC26A4 are associated with Pendred syndrome. Elevated SLC26A4 expression has been linked to airway inflammation, hyperreactivity, and mucus production in asthma. Here, we review novel insights from SLC gene family members into the mechanisms of substrate transport and disease associations, with specific emphasis on SLC26A4. We explore triggers inducing SLC26A4 expression and its contributions to the pathogenesis of pulmonary diseases, particularly asthma. We summarize the inhibitors of SLC26A4 that have shown promise in the treatment of different phenotypes of diseases. While SLC26A4 inhibitors present potential treatments for asthma, further research is imperative to delineate their precise role in asthma pathogenesis and develop efficacious therapeutic strategies targeting this protein.
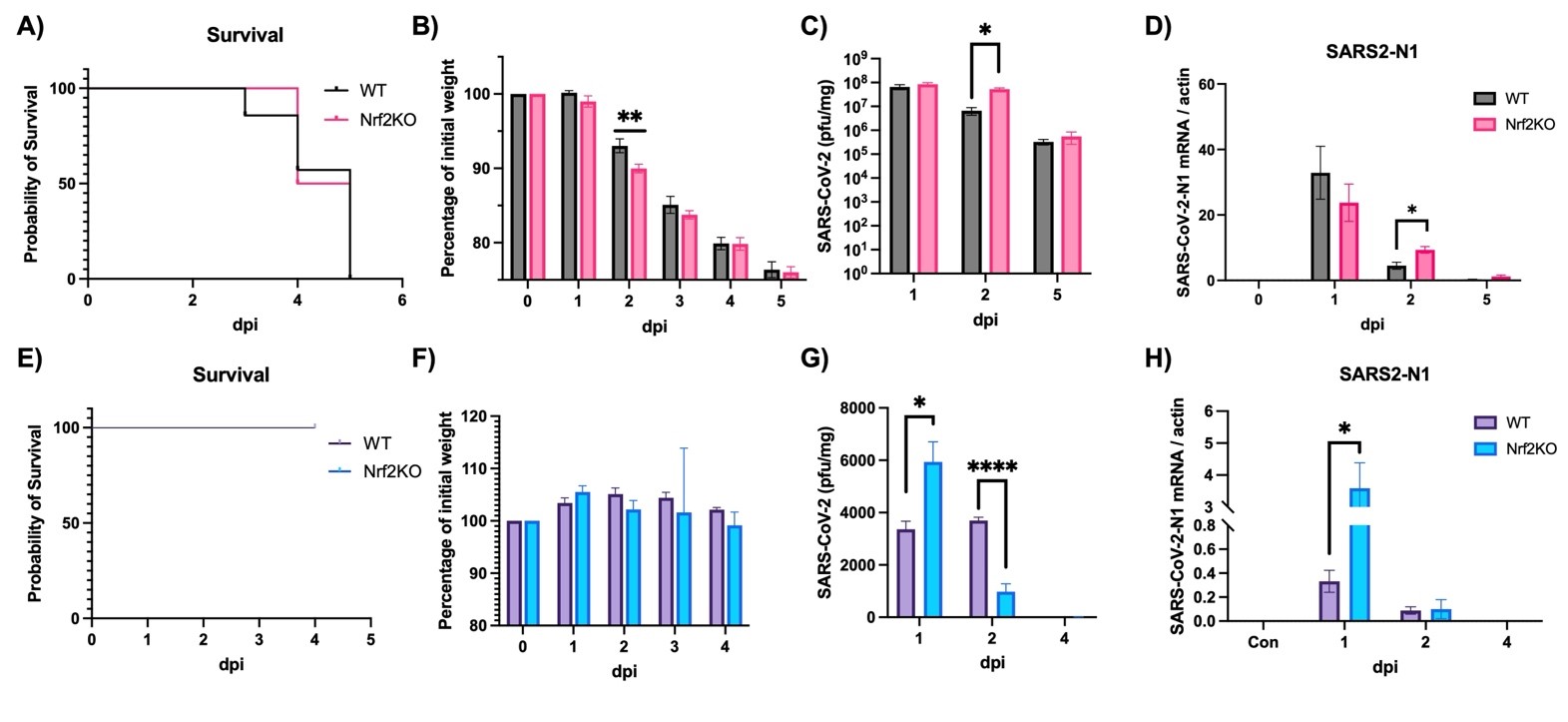
It is well established that Nrf2 plays a crucial role in anti-oxidant and anti-inflammatory functions. However, its antiviral capabilities remain less explored. Despite this, several Nrf2 activators have demonstrated anti-SARS-CoV-2 properties, though the mechanisms behind these effects are not fully understood. In this study, using two mouse models of SARS-CoV-2 infection, we observed that the absence of Nrf2 significantly increased viral load and altered inflammatory responses. Additionally, we evaluated five Nrf2 modulators. Notably, epigallocatechin gallate (EGCG), sulforaphane (SFN), and dimethyl fumarate (DMF) exhibited significant antiviral effects, with SFN being the most effective. SFN did not impact viral entry but appeared to inhibit the main protease (MPro) of SARS-CoV-2, encoded by the Nsp5 gene, as indicated by two protease inhibition assays. Moreover, using two Nrf2 knockout cell lines, we confirmed that SFN's antiviral activity occurs independently of Nrf2 activation in vitro. Paradoxically, in vivo tests using the MA30 model showed that SFN's antiviral function was completely lost in Nrf2 knockout mice. Thus, although SFN and potentially other Nrf2 modulators can inhibit SARS-CoV-2 independently of Nrf2 activation in cell models, their Nrf2-dependent activities might be crucial for antiviral defense under physiological conditions.
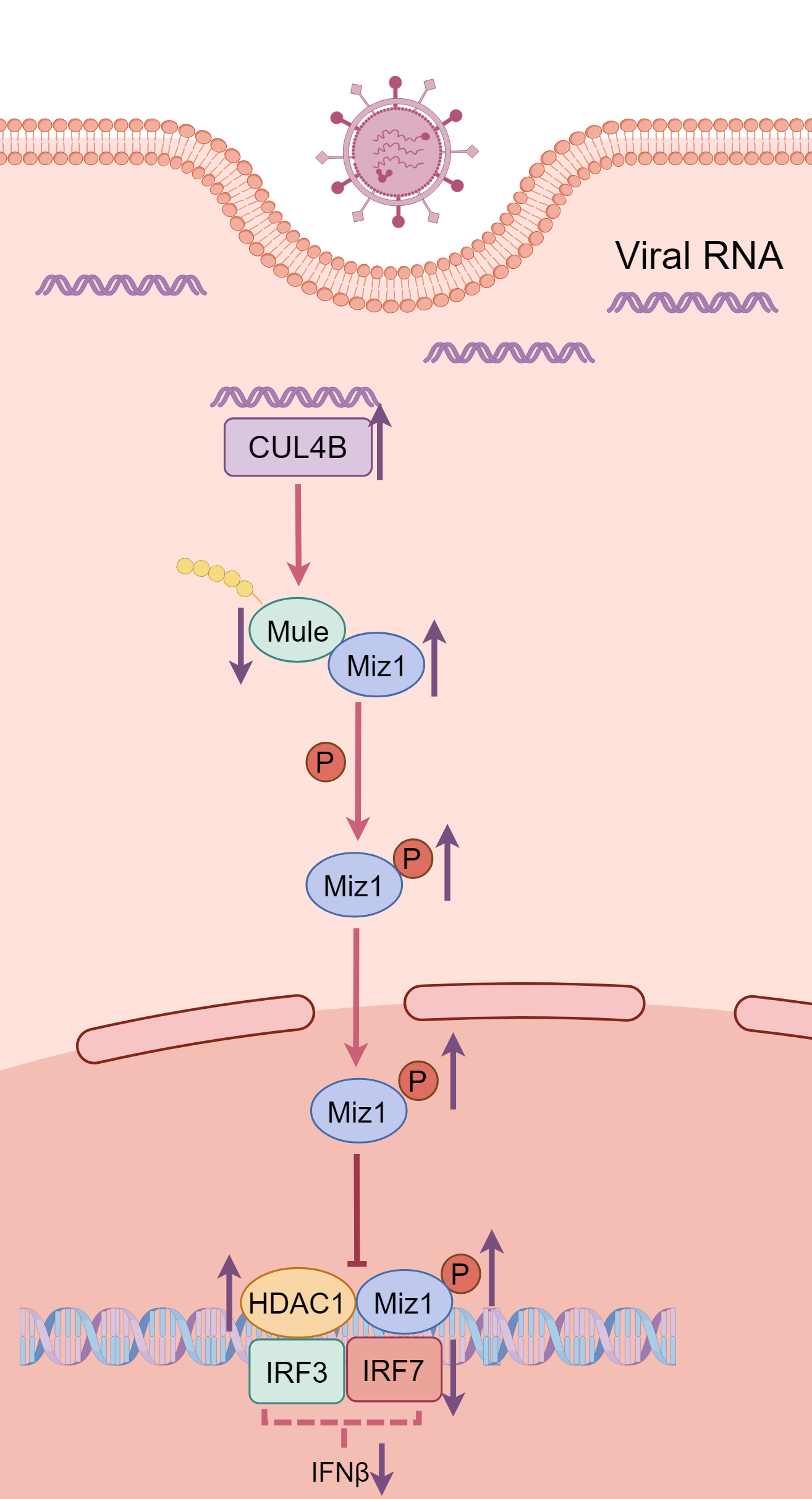
The ubiquitin system has been shown to play an important role in regulation of immune responses during viral infection. In a recent article published in Science Signaling, Wu and colleagues revealed that transcriptional factor Miz1 plays a pro-viral role in influenza A virus (IAV) infection by suppressing type I interferons (IFNs) production through recruiting HDAC1 to ifnb1 promoter. They show that a series of E3 ligases combinatorially regulates Miz1 ubiquitination and degradation and modulates IFNs production and viral replication.
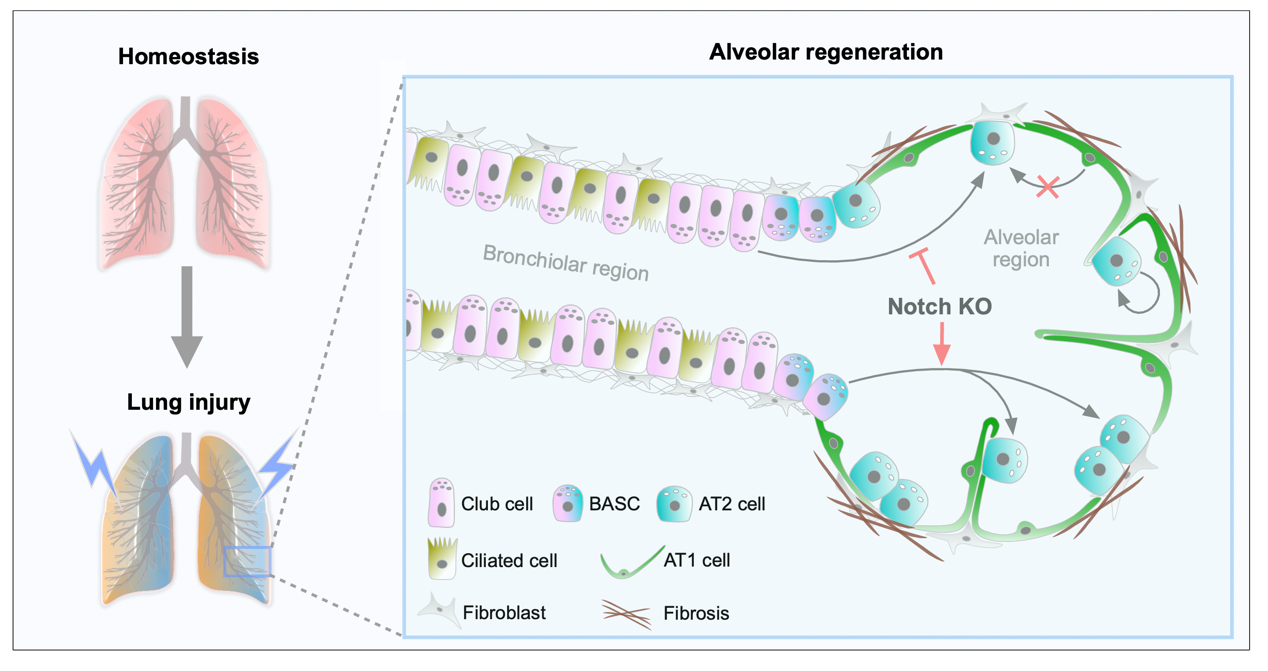
As alveolar epithelial stem cells, alveolar type II (AT2) cells play a pivotal role in sustaining alveolar homeostasis and facilitating repair processes. However, the sources of AT2 cell regeneration have remained contentious due to the non-specific labeling limitations of traditional single recombinase-based lineage tracing techniques. To address this issue, we employed dual recombination systems to develop more precise lineage tracing methodologies, effectively bypassing the shortcomings of conventional approaches and enabling specific labeling of lung epithelial cells. Our findings demonstrate that, following lung injury, regenerated AT2 cells do not originate from alveolar type I (AT1) cells, but instead derive from bronchiolar club cells and bronchioalveolar stem cells (BASCs), alongside the self-renewal of resident AT2 cells. Furthermore, we discovered that the transition of club cells and BASCs into AT2 cells is distinctly modulated by the Notch signaling pathway. This study not only provides novel insights into lung regeneration, but the innovative lineage tracing technology developed herein also holds promise as a technical support for research in diverse fields.
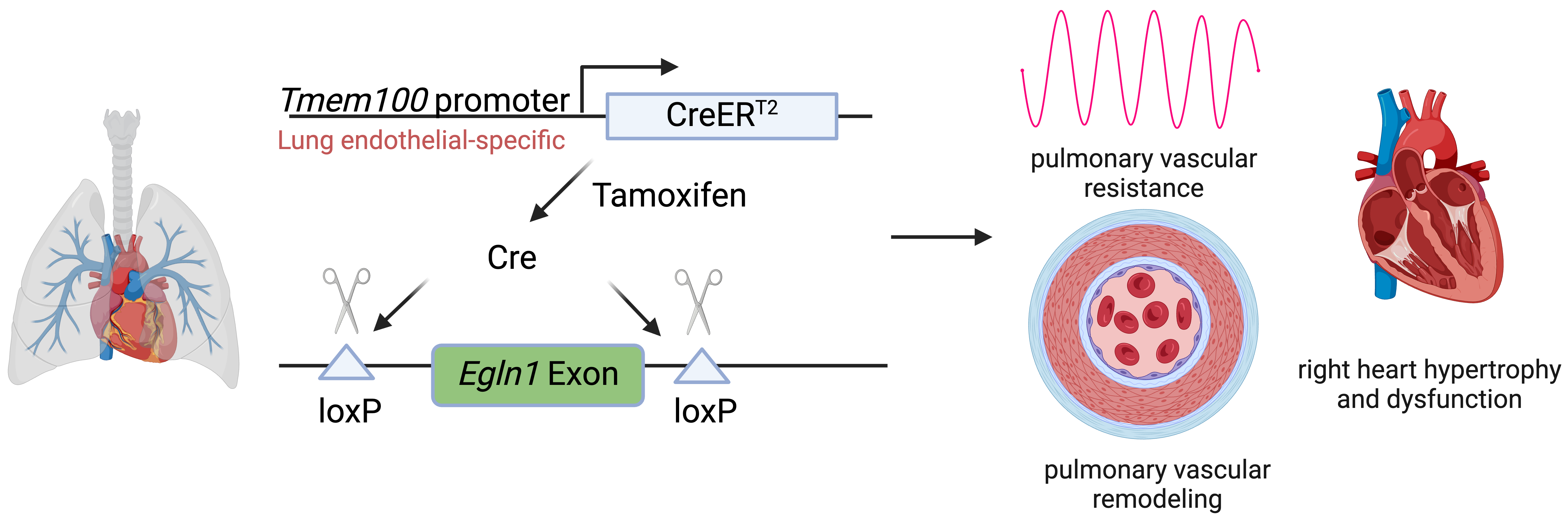
Pulmonary arterial hypertension (PAH) is a devastating disease characterized by high blood pressure in the pulmonary arteries, which can potentially lead to heart failure over time. Previously, our lab found that endothelia-specific knockout of Egln1, encoding prolyl 4-hydroxylase-2 (PHD2), induced spontaneous pulmonary hypertension (PH). Recently, we elucidated that Tmem100 is a lung-specific endothelial gene using Tmem100-CreERT2 mice. We hypothesize that lung endothelial-specific deletion of Egln1 could lead to the development of PH without affecting Egln1 gene expression in other organs. Tmem100-CreERT2 mice were crossed with Egln1flox/flox mice to generate Egln1f/f;Tmem100-CreERT2 (LiCKO) mice. Western blot and immunofluorescent staining were performed to verify the knockout efficacy of Egln1 in multiple organs of LiCKO mice. PH phenotypes, including hemodynamics, right heart size and function, pulmonary vascular remodeling, were evaluated by right heart catheterization and echocardiography measurements. Tamoxifen treatment induced Egln1 deletion in the lung endothelial cells (ECs) but not in other organs of adult LiCKO mice. LiCKO mice exhibited an increase in right ventricular systolic pressure (RVSP, ~35 mmHg) and right heart hypertrophy. Echocardiography measurements showed right heart hypertrophy, as well as cardiac and pulmonary arterial dysfunction. Pulmonary vascular remodeling, including increased pulmonary wall thickness and muscularization of distal pulmonary arterials, was enhanced in LiCKO mice compared to wild-type mice. Tmem100 promoter-mediated lung endothelial knockout of Egln1 in mice leads to development of spontaneous PH. LiCKO mice could serve as a novel mouse model for PH to study lung and other organ crosstalk.